nf-core/rnaseq
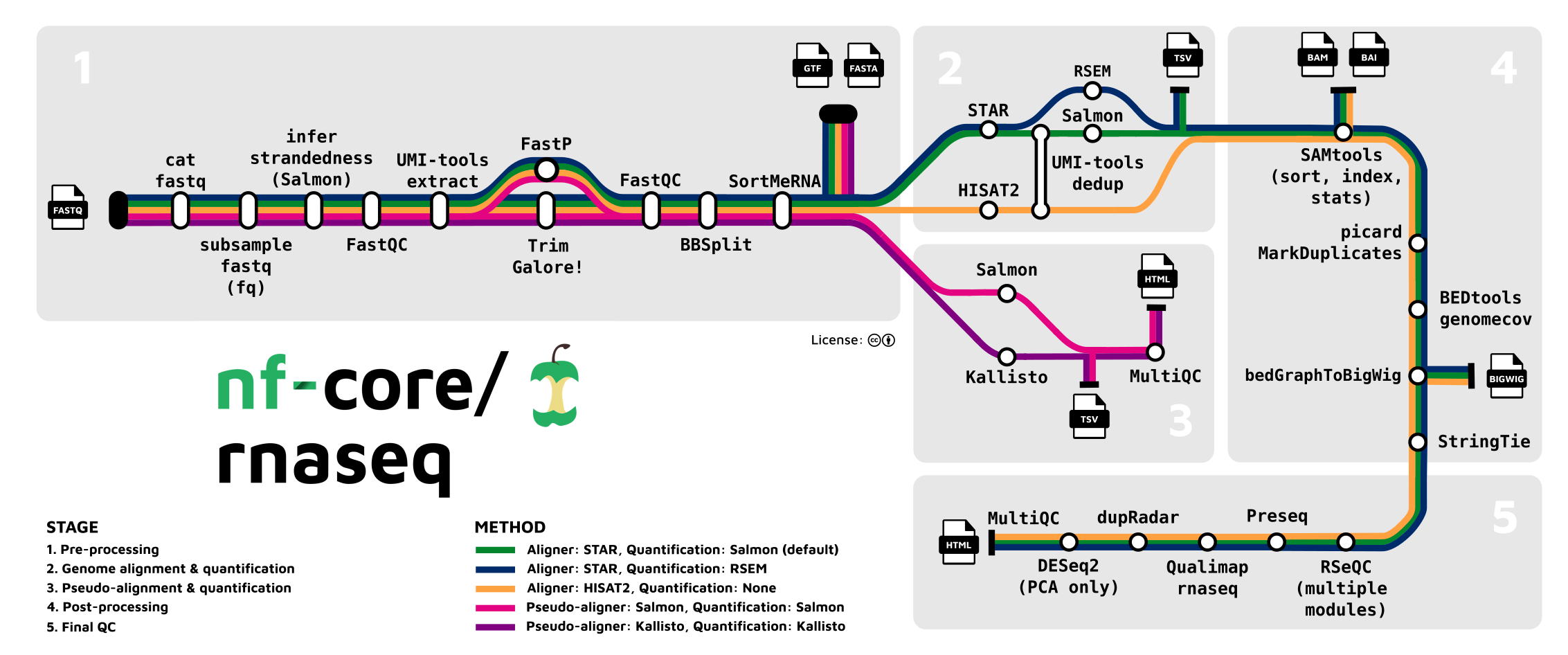
nf-core/rnaseq is a bioinformatics pipeline that can be used to analyse RNA sequencing data obtained from organisms with a reference genome and annotation. It takes a samplesheet and FASTQ files as input, performs quality control (QC), trimming and (pseudo-)alignment, and produces a gene expression matrix and extensive QC report.
Create the working directory
mkdir -p /cluster/tufts/workshop/UTLN/rnaseq ## Replace UTLN with your own UTLN
Prepare the input samplesheet
Samplesheet generated by fetchngs
cd /cluster/tufts/workshop/UTLN/fetchngs/fetchngsOut/samplesheet
ls
You should see the below output:
id_mappings.csv multiqc_config.yml samplesheet.csv
We can use samplesheet.csv directly for RNAseq analysis, but I prefer creating a simpler samplesheet.
head -n 3 samplesheet.csv
"sample","fastq_1","fastq_2","strandedness","run_accession","experiment_accession","sample_accession","secondary_sample_accession","study_accession","secondary_study_accession","submission_accession","run_alias","experiment_alias","sample_alias","study_alias","library_layout","library_selection","library_source","library_strategy","library_name","instrument_model","instrument_platform","base_count","read_count","tax_id","scientific_name","sample_title","experiment_title","study_title","sample_description","fastq_md5","fastq_bytes","fastq_ftp","fastq_galaxy","fastq_aspera"
"SRX1693951","fetchngsOut/fastq/SRX1693951_SRR3362661_1.fastq.gz","fetchngsOut/fastq/SRX1693951_SRR3362661_2.fastq.gz","auto","SRR3362661","SRX1693951","SAMN04639576","SRS1386868","PRJNA318251","SRP073189","SRA409858","GSM2114336_r1","GSM2114336","GSM2114336","GSE80182","PAIRED","cDNA","TRANSCRIPTOMIC","RNA-Seq","","Illumina HiSeq 2500","ILLUMINA","9527214600","31757382","9606","Homo sapiens","A549_GFPkd_1","Illumina HiSeq 2500 sequencing: GSM2114336: A549_GFPkd_1 Homo sapiens RNA-Seq","A TGFbeta-PRMT5-MEP50 Axis Regulates Cancer Cell Invasion through Histone H3 and H4 Arginine Methylation Coupled Transcriptional Activation and Repression","A549_GFPkd_1","d2da6755e6219a091e0a84e9fc63a6fa;ac0e3cd5d59ef55aa3c6d3bb0c31a10c","3720225514;3596832711","ftp.sra.ebi.ac.uk/vol1/fastq/SRR336/001/SRR3362661/SRR3362661_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR336/001/SRR3362661/SRR3362661_2.fastq.gz","ftp.sra.ebi.ac.uk/vol1/fastq/SRR336/001/SRR3362661/SRR3362661_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR336/001/SRR3362661/SRR3362661_2.fastq.gz","fasp.sra.ebi.ac.uk:/vol1/fastq/SRR336/001/SRR3362661/SRR3362661_1.fastq.gz;fasp.sra.ebi.ac.uk:/vol1/fastq/SRR336/001/SRR3362661/SRR3362661_2.fastq.gz"
"SRX1693952","fetchngsOut/fastq/SRX1693952_SRR3362662_1.fastq.gz","fetchngsOut/fastq/SRX1693952_SRR3362662_2.fastq.gz","auto","SRR3362662","SRX1693952","SAMN04639580","SRS1386867","PRJNA318251","SRP073189","SRA409858","GSM2114337_r1","GSM2114337","GSM2114337","GSE80182","PAIRED","cDNA","TRANSCRIPTOMIC","RNA-Seq","","Illumina HiSeq 2500","ILLUMINA","11198386200","37327954","9606","Homo sapiens","A549_GFPkd_2","Illumina HiSeq 2500 sequencing: GSM2114337: A549_GFPkd_2 Homo sapiens RNA-Seq","A TGFbeta-PRMT5-MEP50 Axis Regulates Cancer Cell Invasion through Histone H3 and H4 Arginine Methylation Coupled Transcriptional Activation and Repression","A549_GFPkd_2","152754e5004274d7e0d8f56ea481e2d7;f2290b266571d98c782e7614528ab3c9","4367522067;4230444730","ftp.sra.ebi.ac.uk/vol1/fastq/SRR336/002/SRR3362662/SRR3362662_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR336/002/SRR3362662/SRR3362662_2.fastq.gz","ftp.sra.ebi.ac.uk/vol1/fastq/SRR336/002/SRR3362662/SRR3362662_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR336/002/SRR3362662/SRR3362662_2.fastq.gz","fasp.sra.ebi.ac.uk:/vol1/fastq/SRR336/002/SRR3362662/SRR3362662_1.fastq.gz;fasp.sra.ebi.ac.uk:/vol1/fastq/SRR336/002/SRR3362662/SRR3362662_2.fastq.gz"
Reformating samplesheet
awk -F"," 'OFS=","{print $27,$2,$3,$4}' samplesheet.csv | sed 's/A549_//g' | sed 's/sample_title/sample/g'> /cluster/tufts/workshop/UTLN/rnaseq/samplesheet.csv
We can clearly see that the samplesheet we generated is simple to read. And it only has 4 columns: sample, fastq_1, fastq_2, and strandness.
cat /cluster/tufts/workshop/UTLN/rnaseq/samplesheet.csv
"sample","fastq_1","fastq_2","strandedness"
"GFPkd_1","fetchngsOut/fastq/SRX1693951_SRR3362661_1.fastq.gz","fetchngsOut/fastq/SRX1693951_SRR3362661_2.fastq.gz","auto"
"GFPkd_2","fetchngsOut/fastq/SRX1693952_SRR3362662_1.fastq.gz","fetchngsOut/fastq/SRX1693952_SRR3362662_2.fastq.gz","auto"
"GFPkd_3","fetchngsOut/fastq/SRX1693953_SRR3362663_1.fastq.gz","fetchngsOut/fastq/SRX1693953_SRR3362663_2.fastq.gz","auto"
"PRMT5kd_1","fetchngsOut/fastq/SRX1693954_SRR3362664_1.fastq.gz","fetchngsOut/fastq/SRX1693954_SRR3362664_2.fastq.gz","auto"
"PRMT5kd_2","fetchngsOut/fastq/SRX1693955_SRR3362665_1.fastq.gz","fetchngsOut/fastq/SRX1693955_SRR3362665_2.fastq.gz","auto"
"PRMT5kd_3","fetchngsOut/fastq/SRX1693956_SRR3362666_1.fastq.gz","fetchngsOut/fastq/SRX1693956_SRR3362666_2.fastq.gz","auto"
Note
Please note that the fastq_1 and fastq_2 columns in the data contain the location of fastq files. However, these paths are relative rather than absolute. To ensure the rnaseq pipeline can locate these fastq files, we can create a soft link for fetchngsOut in the working directory of rnaseq pipeline.
cd /cluster/tufts/workshop/UTLN/rnaseq/
ln -s /cluster/tufts/workshop/UTLN/fetchngs/fetchngsOut .
ls -l
Please verify that we have successfully linked fetchngsOut to the current directory.
lrwxrwxrwx 1 yzhang85 workshop 53 Apr 2 18:19 fetchngsOut -> /cluster/tufts/workshop/yzhang85/fetchngs/fetchngsOut/
-rw-rw---- 1 yzhang85 workshop 788 Apr 2 18:18 samplesheet.csv
rnaseq on Open OnDemand
We have already downloaded the raw fastq files for RNAseq using fetchngs. However, for conducting RNAseq analysis, we also need the reference genome fasta file and gtf annotation file. Since these are human samples, we require the human reference genome.
There are two ways to obtain the human reference genome:
- Choose
GRCh38iniGenomes. This method is simple, and theiGenomeshave been set up for users. You only need to select the reference to use. However, this method isnot recommendedbecause the annotation files iniGenomeshave not been updated in some years, making them out of date. We advise against using them for your research and recommend them for classroom/workshop purposes only. - Download the latest version of genomes from public databases such as
EnsemblorNCBI.
In this workshop, we will guide you on how to download your own reference genomes from Ensemble database.
Open OnDemand Arguments
- Number of hours: 24
- Select cpu partition: batch
- Reservation for class, training, workshop: Default
- Version: 3.14.0
- Working Directory:
/cluster/tufts/workshop/UTLN/rnaseq/## Replace UTLN with your own UTLN - outdir: rnaseqOut
- input: samplesheet.csv
- multiqc_title: PRMT5kd vs. GFPkd
- iGenomes: None
- fasta: https://ftp.ensembl.org/pub/release-111/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
- gtf: https://ftp.ensembl.org/pub/release-111/gtf/homo_sapiens/Homo_sapiens.GRCh38.111.gtf.gz
- trimmer: trimgalore
- extra_fastp_args: -q 35 --paired
- aligner: star_salmon
- save_reference: true
- skip_umi_extract: true
- skip_pseudo_alignment: true
- skip_stringtie: true
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/rnaseq v3.14.0
------------------------------------------------------
Core Nextflow options
runName : irreverent_watson
containerEngine : singularity
launchDir : /cluster/tufts/workshop/UTLN/rnaseq
workDir : /cluster/tufts/workshop/UTLN/rnaseq/work
projectDir : /cluster/tufts/biocontainers/nf-core/pipelines/nf-core-rnaseq/3.14.0/3_14_0
userName : yzhang85
profile : tufts
configFiles :
Input/output options
input : samplesheet.csv
outdir : rnaseqOut
multiqc_title : PRMT5kd vs. GFPkd
Reference genome options
fasta : https://ftp.ensembl.org/pub/release-111/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
gtf : https://ftp.ensembl.org/pub/release-111/gtf/homo_sapiens/Homo_sapiens.GRCh38.111.gtf.gz
igenomes_base : /cluster/tufts/biocontainers/datasets/igenomes/
Read trimming options
extra_trimgalore_args : -q 35 --paired
Optional outputs
save_reference : true
Process skipping options
skip_umi_extract : true
skip_pseudo_alignment : true
skip_stringtie : true
Institutional config options
config_profile_description: The Tufts University HPC cluster profile provided by nf-core/configs.
config_profile_contact : Yucheng Zhang
config_profile_url : https://it.tufts.edu/high-performance-computing
Max job request options
max_cpus : 72
max_memory : 120 GB
max_time : 7d
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------
------------------------------------------------------
If you use nf-core/rnaseq for your analysis please cite:
* The pipeline
https://doi.org/10.5281/zenodo.1400710
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/rnaseq/blob/master/CITATIONS.md
[- ] process > NFCORE_RNASEQ:RNASEQ:PREPAR... -
[- ] process > NFCORE_RNASEQ:RNASEQ:PREPAR... -
[- ] process > NFCORE_RNASEQ:RNASEQ:PREPAR... -
[- ] process > NFCORE_RNASEQ:RNASEQ:PREPAR... -
[- ] process > NFCORE_RNASEQ:RNASEQ:PREPAR... -
[- ] process > NFCORE_RNASEQ:RNASEQ:PREPAR... -
[- ] process > NFCORE_RNASEQ:RNASEQ:PREPAR... -
[- ] process > NFCORE_RNASEQ:RNASEQ:PREPAR... -
[- ] process > NFCORE_RNASEQ:RNASEQ:PREPAR... -
[- ] process > NFCORE_RNASEQ:RNASEQ:PREPAR... -
[- ] process > NFCORE_RNASEQ:RNASEQ:PREPAR... -
[- ] process > NFCORE_RNASEQ:RNASEQ:PREPAR... -
[- ] process > NFCORE_RNASEQ:RNASEQ:CAT_FASTQ -
[- ] process > NFCORE_RNASEQ:RNASEQ:FASTQ_... -
[- ] process > NFCORE_RNASEQ:RNASEQ:FASTQ_... -
.
.
.
Monitor the execution with Nextflow Tower using this URL: https://tower.nf/user/yucheng-zhang/watch/5di71eyYto2FH9
executor > slurm (209)
[d1/517f0f] process > NFCORE_RNASEQ:RNASEQ:PREPAR... [100%] 1 of 1 ✔
[29/eb99ef] process > NFCORE_RNASEQ:RNASEQ:PREPAR... [100%] 1 of 1 ✔
[eb/937d34] process > NFCORE_RNASEQ:RNASEQ:PREPAR... [100%] 1 of 1 ✔
[30/ba7437] process > NFCORE_RNASEQ:RNASEQ:PREPAR... [100%] 1 of 1 ✔
[f8/be5fd5] process > NFCORE_RNASEQ:RNASEQ:PREPAR... [100%] 1 of 1 ✔
[b8/092179] process > NFCORE_RNASEQ:RNASEQ:PREPAR... [100%] 1 of 1 ✔
[c1/7d9c49] process > NFCORE_RNASEQ:RNASEQ:PREPAR... [100%] 1 of 1 ✔
[- ] process > NFCORE_RNASEQ:RNASEQ:CAT_FASTQ -
[2e/e95ccb] process > NFCORE_RNASEQ:RNASEQ:FASTQ_... [100%] 6 of 6 ✔
[d3/24f617] process > NFCORE_RNASEQ:RNASEQ:FASTQ_... [100%] 6 of 6 ✔
[97/2bb6ec] process > NFCORE_RNASEQ:RNASEQ:FASTQ_... [100%] 1 of 1 ✔
[49/f116ce] process > NFCORE_RNASEQ:RNASEQ:FASTQ_... [100%] 6 of 6 ✔
[66/7832b0] process > NFCORE_RNASEQ:RNASEQ:FASTQ_... [100%] 6 of 6 ✔
[df/7eb1ea] process > NFCORE_RNASEQ:RNASEQ:ALIGN_... [100%] 6 of 6 ✔
[6a/857d7c] process > NFCORE_RNASEQ:RNASEQ:ALIGN_... [100%] 6 of 6 ✔
[46/65e709] process > NFCORE_RNASEQ:RNASEQ:ALIGN_... [100%] 6 of 6 ✔
[06/684719] process > NFCORE_RNASEQ:RNASEQ:ALIGN_... [100%] 6 of 6 ✔
[01/2fc01d] process > NFCORE_RNASEQ:RNASEQ:ALIGN_... [100%] 6 of 6 ✔
[0f/f9bf31] process > NFCORE_RNASEQ:RNASEQ:ALIGN_... [100%] 6 of 6 ✔
[81/eaf8db] process > NFCORE_RNASEQ:RNASEQ:QUANTI... [100%] 6 of 6 ✔
[d5/12a872] process > NFCORE_RNASEQ:RNASEQ:QUANTI... [100%] 1 of 1 ✔
[17/ceb58b] process > NFCORE_RNASEQ:RNASEQ:QUANTI... [100%] 1 of 1 ✔
[f7/3dcf2d] process > NFCORE_RNASEQ:RNASEQ:QUANTI... [100%] 1 of 1 ✔
[4b/2991d1] process > NFCORE_RNASEQ:RNASEQ:QUANTI... [100%] 1 of 1 ✔
[9d/1204f5] process > NFCORE_RNASEQ:RNASEQ:QUANTI... [100%] 1 of 1 ✔
[14/249750] process > NFCORE_RNASEQ:RNASEQ:QUANTI... [100%] 1 of 1 ✔
[76/2d571a] process > NFCORE_RNASEQ:RNASEQ:DESEQ2... [100%] 1 of 1 ✔
[b7/1658e4] process > NFCORE_RNASEQ:RNASEQ:BAM_MA... [100%] 6 of 6 ✔
[64/31c31b] process > NFCORE_RNASEQ:RNASEQ:BAM_MA... [100%] 6 of 6 ✔
[c6/9ff3a9] process > NFCORE_RNASEQ:RNASEQ:BAM_MA... [100%] 6 of 6 ✔
[36/df1dd9] process > NFCORE_RNASEQ:RNASEQ:BAM_MA... [100%] 6 of 6 ✔
[f4/fb9e0d] process > NFCORE_RNASEQ:RNASEQ:BAM_MA... [100%] 6 of 6 ✔
[42/28fcdb] process > NFCORE_RNASEQ:RNASEQ:SUBREA... [100%] 6 of 6 ✔
[68/b06f5c] process > NFCORE_RNASEQ:RNASEQ:MULTIQ... [100%] 6 of 6 ✔
[ce/7d55f5] process > NFCORE_RNASEQ:RNASEQ:BEDTOO... [100%] 6 of 6 ✔
[24/45057e] process > NFCORE_RNASEQ:RNASEQ:BEDGRA... [100%] 6 of 6 ✔
[36/194966] process > NFCORE_RNASEQ:RNASEQ:BEDGRA... [100%] 6 of 6 ✔
[de/70fc07] process > NFCORE_RNASEQ:RNASEQ:BEDGRA... [100%] 6 of 6 ✔
[be/a747c3] process > NFCORE_RNASEQ:RNASEQ:BEDGRA... [100%] 6 of 6 ✔
[2b/25dca2] process > NFCORE_RNASEQ:RNASEQ:QUALIM... [100%] 6 of 6 ✔
[21/ebcfd0] process > NFCORE_RNASEQ:RNASEQ:DUPRAD... [100%] 6 of 6 ✔
[91/91276e] process > NFCORE_RNASEQ:RNASEQ:BAM_RS... [100%] 6 of 6 ✔
[18/b555d7] process > NFCORE_RNASEQ:RNASEQ:BAM_RS... [100%] 6 of 6 ✔
[8c/a1307b] process > NFCORE_RNASEQ:RNASEQ:BAM_RS... [100%] 6 of 6 ✔
[5f/d7cac7] process > NFCORE_RNASEQ:RNASEQ:BAM_RS... [100%] 6 of 6 ✔
[c7/ce1ec3] process > NFCORE_RNASEQ:RNASEQ:BAM_RS... [100%] 6 of 6 ✔
[78/10ee50] process > NFCORE_RNASEQ:RNASEQ:BAM_RS... [100%] 6 of 6 ✔
[44/7c4656] process > NFCORE_RNASEQ:RNASEQ:BAM_RS... [100%] 6 of 6 ✔
[56/57e6ac] process > NFCORE_RNASEQ:RNASEQ:CUSTOM... [100%] 1 of 1 ✔
[6b/f6514d] process > NFCORE_RNASEQ:RNASEQ:MULTIQ... [100%] 1 of 1 ✔
-[nf-core/rnaseq] Pipeline completed successfully -
Completed at: 26-Mar-2024 14:57:46
Duration : 6h 45m 41s
CPU hours : 109.2
Succeeded : 209
Cleaning up...
Running pipeline on the command line
If you prefer to run the pipelines using the command line interface, you can submit a slurm jobscript with the following code.
Run the pipeline directly
module load nf-core
export NXF_SINGULARITY_CACHEDIR=/cluster/tufts/biocontainers/nf-core/singularity-images
nextflow run /cluster/tufts/biocontainers/nf-core/pipelines/nf-core-rnaseq/3.14.0/3_14_0
-profile tufts \
--input samplesheet.csv \
--outdir rnaseqOut \
--gtf "https://ftp.ensembl.org/pub/release-111/gtf/homo_sapiens/Homo_sapiens.GRCh38.111.gtf.gz" \
--fasta "https://ftp.ensembl.org/pub/release-111/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz" \
--extra_trimgalore_args "-q 35 --paired" \
--skip_pseudo_alignment \
--save_reference
Another easier way
module load nf-core-rnaseq/3.14.0
rnaseq -profile tufts \
--input samplesheet.csv \
--outdir rnaseqOut \
--gtf "https://ftp.ensembl.org/pub/release-111/gtf/homo_sapiens/Homo_sapiens.GRCh38.111.gtf.gz" \
--fasta "https://ftp.ensembl.org/pub/release-111/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz" \
--extra_trimgalore_args "-q 35 --paired" \
--skip_pseudo_alignment \
--save_reference
Nextflow clean
Clean the work
You can clean the work directory, by mannualy run
rm -rf work