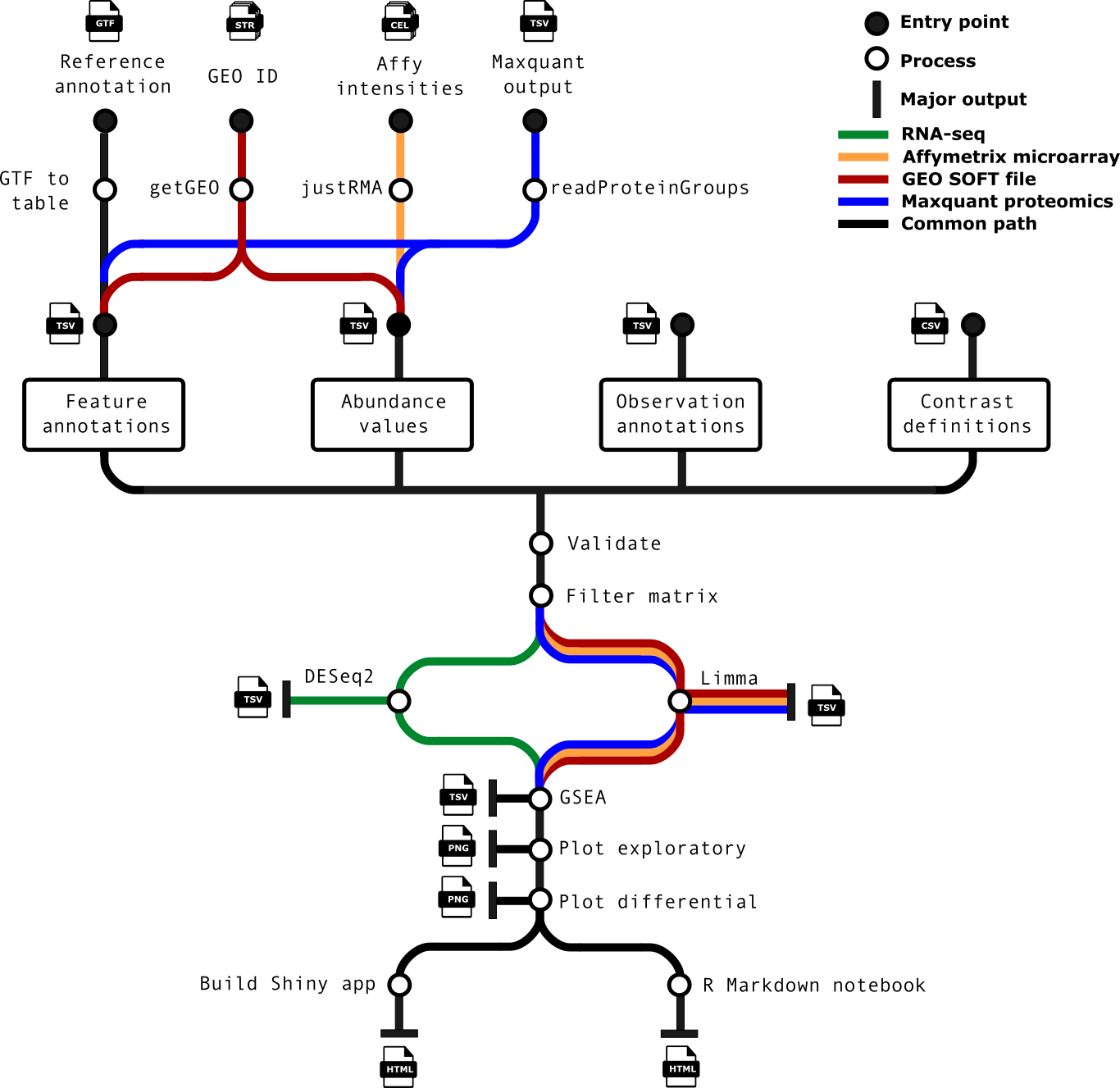
nf-core/differentialabundance
nf-core/differentialabundance is a bioinformatics pipeline that can be used to analyse data represented as matrices, comparing groups of observations to generate differential statistics and downstream analyses. The pipeline supports RNA-seq data such as that generated by the nf-core rnaseq workflow, and Affymetrix arrays via .CEL files.
Create the working directory
mkdir -p /cluster/tufts/workshop/UTLN/differentialabundance
reference genome gtf
In the last RNAseq workshop, we selected save_reference. So that all refereneced data will be saved for our future use. Today we can reuse the gtf file for human genome.
ls -1 /cluster/tufts/workshop/UTLN/rnaseq/rnaseqOut/genome/
You can see the GRCh38 reference genome's gtf and fasta files. In addition, you can see the newly created STAR index and rsem folders that can be used for your future RNA-Seq analysis.
Homo_sapiens.GRCh38.111.gtf
Homo_sapiens.GRCh38.dna.primary_assembly.fa
Homo_sapiens.GRCh38.dna.primary_assembly.fa.fai
Homo_sapiens.GRCh38.dna.primary_assembly.fa.sizes
Homo_sapiens.GRCh38.dna.primary_assembly.filtered.bed
Homo_sapiens.GRCh38.dna.primary_assembly.filtered.gtf
genome.transcripts.fa
index/
rsem/
Let's create a softlink of the gtf to our differentialabundance folder
cd /cluster/tufts/workshop/UTLN/differentialabundance
ln -s /cluster/tufts/workshop/UTLN/rnaseq/rnaseqOut/genome/Homo_sapiens.GRCh38.111.gtf .
gene expression count matrix
In the output folder of RNAseq workshop, you can find the count file we need salmon.merged.gene_counts.tsv via ls.
$ ls -1 /cluster/tufts/workshop/UTLN/rnaseq/rnaseqOut/star_salmon/*.tsv
/cluster/tufts/workshop/UTLN/rnaseq/rnaseqOut/star_salmon/salmon.merged.gene_counts.tsv
/cluster/tufts/workshop/UTLN/rnaseq/rnaseqOut/star_salmon/salmon.merged.gene_counts_length_scaled.tsv
/cluster/tufts/workshop/UTLN/rnaseq/rnaseqOut/star_salmon/salmon.merged.gene_counts_scaled.tsv
/cluster/tufts/workshop/UTLN/rnaseq/rnaseqOut/star_salmon/salmon.merged.gene_lengths.tsv
/cluster/tufts/workshop/UTLN/rnaseq/rnaseqOut/star_salmon/salmon.merged.gene_tpm.tsv
/cluster/tufts/workshop/UTLN/rnaseq/rnaseqOut/star_salmon/salmon.merged.transcript_counts.tsv
/cluster/tufts/workshop/UTLN/rnaseq/rnaseqOut/star_salmon/salmon.merged.transcript_lengths.tsv
/cluster/tufts/workshop/UTLN/rnaseq/rnaseqOut/star_salmon/salmon.merged.transcript_tpm.tsv
/cluster/tufts/workshop/UTLN/rnaseq/rnaseqOut/star_salmon/tx2gene.tsv
We can create a soft link of salmon.merged.gene_counts.tsv into our differentialabundance folder
cd /cluster/tufts/workshop/UTLN/differentialabundance
ln -s /cluster/tufts/workshop/UTLN/rnaseq/rnaseqOut/star_salmon/salmon.merged.gene_counts.tsv .
samplesheet.csv
| sample | treatment | replicate | batch |
|---|---|---|---|
| GFPkd_1 | GFPkd | 1 | A |
| GFPkd_2 | GFPkd | 2 | A |
| GFPkd_3 | GFPkd | 3 | A |
| PRMT5kd_1 | PRMT5kd | 1 | A |
| PRMT5kd_2 | PRMT5kd | 2 | A |
| PRMT5kd_3 | PRMT5kd | 3 | A |
You can copy my samplesheet.csv to your workding directory.
cd /cluster/tufts/workshop/UTLN/differentialabundance
cp /cluster/tufts/workshop/shared/samplesheet.csv .
contrast.csv
| id | variable | reference | target | blocking |
|---|---|---|---|---|
| PRMT5kd_vs_GFPkd | treatment | GFPkd | PRMT5kd |
You can copy my contrast.csv to your working directory.
cd /cluster/tufts/workshop/UTLN/differentialabundance
cp /cluster/tufts/workshop/shared/contrast.csv .
Open OnDemand
Click differentialabundance in Bioinformatics Apps.
Arguments
- Number of hours: 2
- Select cpu parition: batch
- Reservation for class, training, workshop: Bioinformatics Workshop
- Version: 1.4.0
- Working Directory:
/cluster/tufts/workshop/UTLN/differentialabundance## Change this to your own directory - outdir: DEGout
- study_type: rnaseq
- input: samplesheet.csv
- contrasts: contrast.csv
- matrix: salmon.merged.gene_counts.tsv
- observations_id_col: sample
- observations_name_col: sample
- differential_min_fold_change: 1.5
- deseq2_vs_method: rlog
- gsea_run: true
- gsea_gene_sets: /cluster/tufts/workshop/shared/gsea/h.all.v2023.2.Hs.symbols.gmt.txt
- shinyngs_build_app: true
- report_title: PRMT5kd vs. GFPkd
- report_author: Yucheng Zhang ## You can put your name as the author
- gtf: Homo_sapiens.GRCh38.111.gtf
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/differentialabundance v1.4.0
------------------------------------------------------
Core Nextflow options
runName : maniac_mcclintock
containerEngine : singularity
container : [RMARKDOWNNOTEBOOK:biocontainers/r-shinyngs:1.8.4--r43hdfd78af_0]
launchDir : /cluster/tufts/workshop/UTLN/differentialabundance
workDir : /cluster/tufts/workshop/UTLN/differentialabundance/work
projectDir : /cluster/tufts/biocontainers/nf-core/pipelines/nf-core-differentialabundance/1.4.0/1_4_0
userName : yzhang85
profile : tufts
configFiles :
Input/output options
input : samplesheet.csv
contrasts : contrast.csv
outdir : DEGout
Abundance values
matrix : salmon.merged.gene_counts.tsv
affy_cel_files_archive : null
querygse : null
Affy input options
affy_cdfname : null
Differential analysis
differential_min_fold_change: 1.5
Limma specific options (microarray only)
limma_spacing : null
limma_block : null
limma_correlation : null
GSEA
gsea_run : true
gsea_gene_sets : /cluster/tufts/workshop/shared/gsea/h.all.v2023.2.Hs.symbols.gmt.txt
Shiny app settings
shinyngs_shinyapps_account : null
shinyngs_shinyapps_app_name : null
Reporting options
report_file : /cluster/tufts/biocontainers/nf-core/pipelines/nf-core-differentialabundance/1.4.0/1_4_0/assets/differentialabundance_report.Rmd
logo_file : /cluster/tufts/biocontainers/nf-core/pipelines/nf-core-differentialabundance/1.4.0/1_4_0/docs/images/nf-core-differentialabundance_logo_light.png
css_file : /cluster/tufts/biocontainers/nf-core/pipelines/nf-core-differentialabundance/1.4.0/1_4_0/assets/nf-core_style.css
citations_file : /cluster/tufts/biocontainers/nf-core/pipelines/nf-core-differentialabundance/1.4.0/1_4_0/CITATIONS.md
report_title : PRMT5kd vs. GFPkd
report_author : Yucheng Zhang
report_description : null
Institutional config options
config_profile_description : The Tufts University HPC cluster profile provided by nf-core/configs.
config_profile_contact : Yucheng Zhang
config_profile_url : https://it.tufts.edu/high-performance-computing
Max job request options
max_cpus : 72
max_memory : 120 GB
max_time : 7d
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------
If you use nf-core/differentialabundance for your analysis please cite:
* The pipeline
https://doi.org/10.5281/zenodo.7568000
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/differentialabundance/blob/master/CITATIONS.md
[- ] process > NFCORE_DIFFERENTIALABUNDANC... -
[- ] process > NFCORE_DIFFERENTIALABUNDANC... -
[- ] process > NFCORE_DIFFERENTIALABUNDANC... -
[- ] process > NFCORE_DIFFERENTIALABUNDANC... -
[- ] process > NFCORE_DIFFERENTIALABUNDANC... -
[- ] process > NFCORE_DIFFERENTIALABUNDANC... -
[- ] process > NFCORE_DIFFERENTIALABUNDANC... -
[- ] process > NFCORE_DIFFERENTIALABUNDANC... -
[- ] process > NFCORE_DIFFERENTIALABUNDANC... -
[- ] process > NFCORE_DIFFERENTIALABUNDANC... -
[- ] process > NFCORE_DIFFERENTIALABUNDANC... -
[- ] process > NFCORE_DIFFERENTIALABUNDANC... -
.
.
.
executor > slurm (14)
[3c/aa1431] process > NFCORE_DIFFERENTIALABUNDANC... [100%] 1 of 1 ✔
[73/374104] process > NFCORE_DIFFERENTIALABUNDANC... [100%] 1 of 1 ✔
[64/cc51c4] process > NFCORE_DIFFERENTIALABUNDANC... [100%] 1 of 1 ✔
[c8/7b9eb7] process > NFCORE_DIFFERENTIALABUNDANC... [100%] 1 of 1 ✔
[bf/0ac8f6] process > NFCORE_DIFFERENTIALABUNDANC... [100%] 1 of 1 ✔
[f3/85ca6e] process > NFCORE_DIFFERENTIALABUNDANC... [100%] 1 of 1 ✔
[1c/37c98b] process > NFCORE_DIFFERENTIALABUNDANC... [100%] 1 of 1 ✔
[3b/8585ca] process > NFCORE_DIFFERENTIALABUNDANC... [100%] 1 of 1 ✔
[12/f7dac7] process > NFCORE_DIFFERENTIALABUNDANC... [100%] 1 of 1 ✔
[c3/a75051] process > NFCORE_DIFFERENTIALABUNDANC... [100%] 1 of 1 ✔
[3f/7f671c] process > NFCORE_DIFFERENTIALABUNDANC... [100%] 1 of 1 ✔
[51/a37574] process > NFCORE_DIFFERENTIALABUNDANC... [100%] 1 of 1 ✔
[21/f74ad3] process > NFCORE_DIFFERENTIALABUNDANC... [100%] 1 of 1 ✔
[28/fbb019] process > NFCORE_DIFFERENTIALABUNDANC... [100%] 1 of 1 ✔
-[nf-core/differentialabundance] Pipeline completed successfully-
Completed at: 27-Mar-2024 14:02:23
Duration : 25m 13s
CPU hours : 0.5
Succeeded : 14
Cleaning up...
Check the output files
Under the output folder, you will see subfolders listed as below:
other
shinyngs_app
tables
plots
report
pipeline_info
report folder, you will see a html file which will be the report file. Under
shinyngs_app/ folder, you will see a subfolder which stores the app.R shiny app for interactive visualization. You can then view app.R with Open OnDemand shinyngs app.