Approximate time: 30 minutes
Goals
- Align short reads to a references genome
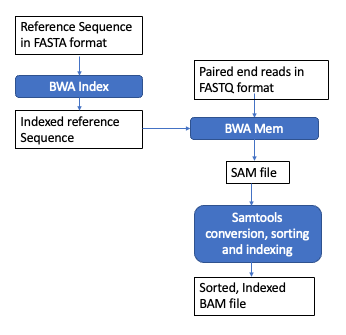
Burrows-Wheeler Aligner (BWA) Overview
BWA is a software package for mapping low-divergent sequences against a large reference genome, such as the human genome. The naive approach to read alignment is to compare a read to every position in the reference genome until a good match is found is far too slow. BWA solves this problem by creating an “index” of our reference sequence for faster lookup.
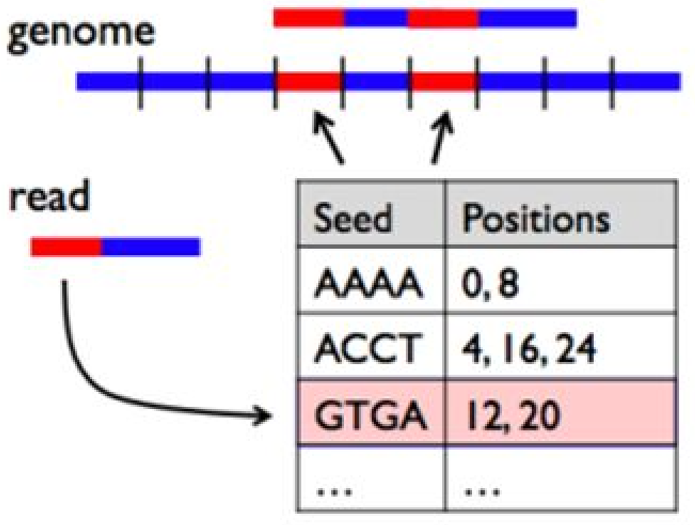
The following figure shows a short read with a red segment followed by a blue segment that we seek to align to a genome containing many blue and red segments. The table keeps track of all the locations where a given pattern of red and blue segments (seed sequence) occurs in the reference genome. When BWA encounters a new read, it looks up the seed sequence at the beginning of the read in the table and retrieves a set of positions that are potential alignment positions for that read. This speeds up the search by reducing the number of positions to check for a good match.
BWA has three algorithms:
- BWA-backtrack: designed for Illumina sequence reads up to 100bp
- BWA-SW: designed for longer sequences ranging from 70bp to 1Mbp, long-read support and split alignment
- BWA-MEM: optimized for 70-100bp Illumina reads
We’ll use BWA-MEM. Underlying the BWA index is the Burrows-Wheeler Transform Video and lecture. This is beyond the scope of this course but is an widely used data compression applications.
Index the reference genome
In the following steps we’ll create the BWA index for the reference genome.
Change to our data directory:
cd ~/hpcDay2/data/
Preview our genome using the command head by typing:
head GCF_009858895.2_ASM985889v3_genomic.fna
You’ll see the first 10 lines of the file which, as discussed, is an example of FASTA format:
>NC_045512.2 Severe acute respiratory syndrome coronavirus... <-- '>' charachter followed by sequence name
ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGTAGAT... <-- sequence
…
Load the BWA module, which will give us access to the bwa program:
module load bwa/0.7.17
Test it out without any arguments in order to view the help message.
bwa
Result:
Program: bwa (alignment via Burrows-Wheeler transformation)
Version: 0.7.17-r1198-dirty
Contact: Heng Li <lh3@sanger.ac.uk>
Usage: bwa <command> [options]
Command: index index sequences in the FASTA format
…
Use the bwa index command to see usage instructions for genome indexing
bwa index
Result:
Usage: bwa index [options] <in.fasta>
Options: -a STR BWT construction algorithm …
Run the command as instructed, using the default options:
bwa index GCF_009858895.2_ASM985889v3_genomic.fna
Result:
[bwa_index] Pack FASTA... 0.00 sec
[bwa_index] Construct BWT for the packed sequence...
[bwa_index] 0.00 seconds elapse.
[bwa_index] Update BWT... 0.00 sec
[bwa_index] Pack forward-only FASTA... 0.00 sec
[bwa_index] Construct SA from BWT and Occ... 0.00 sec
[main] Version: 0.7.9a-r786
[main] CMD: bwa index GCF_009858895.2_ASM985889v3_genomic.fna
[main] Real time: 0.130 sec; CPU: 0.010 sec
When it’s done, take a look at the files produced by typing ls.
The following is the result, with arrows and text on the right giving an explanation of each file.
GCF_009858895.2_ASM985889v3_genomic.fna <-- Original sequence
GCF_009858895.2_ASM985889v3_genomic.fna.amb <-- Location of ambiguous (non-ATGC) nucleotides
GCF_009858895.2_ASM985889v3_genomic.fna.ann <-- Sequence names, lengths
GCF_009858895.2_ASM985889v3_genomic.fna.bwt <-- BWT suffix array
GCF_009858895.2_ASM985889v3_genomic.fna.pac <-- Binary encoded sequence
GCF_009858895.2_ASM985889v3_genomic.fna.sa <-- Suffix array index
BWA alignment
Let’s check the usage instructions for BWA mem by typing bwa mem
Usage: bwa mem [options] <idxbase> <in1.fq> [in2.fq]
Algorithm options:
-t INT number of threads [1]
-k INT minimum seed length [19]
-w INT band width for banded alignment [100]
-d INT off-diagonal X-dropoff [100]
-r FLOAT look for internal seeds inside a seed longer than {-k} * FLOAT [1.5]
-y INT seed occurrence for the 3rd round seeding [20]
-c INT skip seeds with more than INT occurrences [500]
-D FLOAT drop chains shorter than FLOAT fraction of the longest overlapping chain [0.50]
-W INT discard a chain if seeded bases shorter than INT [0]
-m INT perform at most INT rounds of mate rescues for each read [50]
-S skip mate rescue
-P skip pairing; mate rescue performed unless -S also in use
...
Since our alignment command will have multiple arguments, it will be convenient to write a script.
Change into our scripts directory:
cd ../scripts
Open a text editor with the program nano and create a new file called bwa.sh.
nano bwa.sh
Enter the following text.
Note that each line ends in a single backslash \, which will be read as a line continuation.
Be careful to put a space before the backslash and not after.
This serves to make the script more readable.
#!/bin/bash
#SBATCH --job-name=bwa # Job name
#SBATCH --nodes=1 # Nodes requested
#SBATCH -n 2 # Tasks requested
#SBATCH --partition=batch # Parition
#SBATCH --reservation=bioworkshop # Omit this line if not part of workshop
#SBATCH --mem=1Gb # Memory requested
#SBATCH --time=0-30:00 # Time requested
#SBATCH --output=%j.out # Output log file labeled by job name
#SBATCH --error=%j.err # Output error file labeled by job name
# Load the module
module load bwa/0.7.17
# Write the BWA command
bwa mem \
-t 2 \
-o ~/hpcDay2/results/sarscov2.sam \
~/hpcDay2/data/GCF_009858895.2_ASM985889v3_genomic.fna \
~/hpcDay2/data/fastq/trim_galore/SRR15607266_pass_1_val_1.fq.gz \
~/hpcDay2/data/fastq/trim_galore/SRR15607266_pass_2_val_2.fq.gz
Let’s look line by line at the options we’ve given to BWA:
-
-t 2: BWA runs two parallel threads. Alignment is a task that is easy to parallelize because alignment of a read is independent of other reads. Recall that in Setup we asked for a compute node allocation with--cpus=4, which can process up to 8 threads. Here we are using only 2 threads. -
-o results/sarscov2.sam: Place the output in the results folder and give it a name -
The following arguments are the full paths of our reference, read1 and read2 files, in the order required by BWA:
~/hpcDay2/data/GCF_009858895.2_ASM985889v3_genomic.fna \ ~/hpcDay2/data/fastq/trim_galore/SRR15607266_pass_1_val_1.fq.gz \ ~/hpcDay2/data/fastq/trim_galore/SRR15607266_pass_2_val_2.fq.gz
Exit nano by typing ^X and follow prompts to save and name the file bwa.sh.
Now we can run our script.
sbatch bwa.sh
We can check to see if our job is running
squeue -u tutln01
| JOBID | PARTITON | NAME | USER | ST | TIME | NODES | NODELIST(REASON) |
|---|---|---|---|---|---|---|---|
| 24130535 | batch | bwa | tutln01 | R | 0.06 | 1 | c1cmp044 |
| 24129538 | batch | bash | tutln01 | R | 23:14 | 1 | c1cmp044 |
I can see my job number is 24130535. Find your job number.
Take a look at the error and the output files:
ls
bwa.sh 24130535.err 24130535.out
Sequence Alignment Map (SAM)
While our job runs for 3 minutes, we will introduce the SAM format. SAM files have two sections, Header and Alignment.
Header:
@SQ SN:NC_045512.2 LN:29903 <-- Reference sequence name (SN) and length (LN)
@PG ID:bwa PN:bwa VN:0.7.17-r1198-dirty CL:bwa mem -t 2 ... <-- Programs and arguments used in processing
Alignment:
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|
| SRR15607266.1 | 99 | NC_045512.2 | 8152 | 60 | 76M | = | 8307 | 231 | NTTA… | #8ACC… |
| SRR15607266.1 | 147 | NC_045512.2 | 8307 | 60 | 76M | = | 81523 | -231 | AAAA… | GGGG… |
| SRR15607266.2 | 83 | NC_045512.2 | 16369 | 60 | 74M | = | 16255 | -188 | GTTA… | GGFD… |
The fields:
- Read ID
- Flag: indicates alignment information e.g. paired, aligned, etc. Here is a useful site to decode flags.
- Reference sequence name
- Position on the reference sequence where mapping starts
- Mapping Quality
- CIGAR string: summary of alignment, e.g. match (M), insertion (I), deletion (D)
- RNEXT: Name of reference sequence where the other read in the pair aligns
- PNEXT: Position in the reference sequence where the other read in the pair aligns
- TLEN: Template length, size of the original DNA or RNA fragment
- Read Sequence
- Read Quality
More information on SAM format.
Your job should be done, and do do this, check to make sure your bwa job is no longer listed in your squeue -u tutln01 output.
You can also check your resource usage efficiency by running seff. Did you use everything you requested?
Change into our results directory:
cd ../results
List the files in the results directory by typing ls.
Result:
sarscov2.sam
Let’s load the tool Samtools which we’ll use to manipulate SAM files.
To load the module:
module load samtools/1.9
We can view the alignment header with this command:
samtools view -H sarscov2.sam
@SQ SN:NC_045512.2 LN:29903 <-- Reference sequence name (SN) and length (LN)
@PG ID:bwa PN:bwa VN:0.7.17-r1198-dirty CL:bwa mem -t 2 ... <-- Programs and arguments used in processing
We can preview the alignment with this command. We use the bash ‘|’ symbol, called a pipe, which takes the input of one process and passes it as input to another process:
samtools view sarscov2.sam | head
Alignment:
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|
| SRR15607266.1 | 145 | NC_045512.2 | 2 | 60 | 30S42M4S | = | 8365 | 8323 | CGTA… | GGGG… |
| SRR15607266.1 | 97 | NC_045512.2 | 3 | 60 | 32S43M | = | 28292 | 8365 | CCAA… | CCCC… |
| SRR15607266.2 | 161 | NC_045512.2 | 3 | 60 | 32S43M | = | 28255 | 28365 | CCAA… | CCCC… |
Next, we’ll convert the SAM into a compressed, binary format called BAM in order to process it further.
samtools view -bo sarscov2.bam sarscov2.sam
We’ve used options -bo to specify BAM output to a specific file.
‘BAM` files are not human readable, so we use samtools to view:
samtools view sarscov2.bam | head
Sort SAM file
Downstream applications require that reads in SAM files be sorted by reference genome coordinates (fields 3 and 4 in each line of our SAM file). This will assist in fast search, display and other functions.
samtools sort -o sarscov2.srt.bam sarscov2.bam
Where again we’ve used option -o to specify the output file.
Look at the new file, do you notice anything different about the read order?
samtools view sarscov2.srt.bam | head
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|
| SRR15607266.1 | 99 | NC_045512.2 | 8152 | 60 | 76M | = | 8307 | 231 | NTTA… | #8ACC… |
| SRR15607266.1 | 147 | NC_045512.2 | 8307 | 60 | 76M | = | 81523 | -231 | AAAA… | GGGG… |
| SRR15607266.2 | 83 | NC_045512.2 | 16369 | 60 | 74M | = | 16255 | -188 | GTTA… | GGFD… |
Finally, we create a BAM index to speed up searching in downstream applications.
samtools index sarscov2.srt.bam
Alignment Quality Control
Before we view our reads, we’d like to calculate some summary statistics to know how well our reads aligned to the reference genome.
This can be done by running the samtools flagstat program on our sorted BAM file:
samtools flagstat sarscov2.srt.bam
Result:
2583381 + 0 in total (QC-passed reads + QC-failed reads) <-- We have only QC pass reads
58229 + 0 secondary <-- 58229 reads have >1 alignment position
0 + 0 supplementary <-- for reads that align to multiple chromosomes
0 + 0 duplicates
2029614 + 0 mapped (78.56% : N/A) <-- For RNAseq data, >75% is expected,
2525152 + 0 paired in sequencing in this case we have some human contamination
1262576 + 0 read1
1262576 + 0 read2
1948534 + 0 properly paired (77.17% : N/A)
1969890 + 0 with itself and mate mapped
1495 + 0 singletons (0.06% : N/A)
0 + 0 with mate mapped to a different chr
0 + 0 with mate mapped to a different chr (mapQ>=5)
Samtools flagstat is a great way to check to make sure that the alignment meets the quality expected.
In this case, >75% properly paired and mapped indicates a high quality alignment.
Summary