Approximate time: 10 minutes
Learning Objectives
- View alignment, variants and reads using Integrative Genomics Viewer
BAM Visualization with IGV
- With a Chrome web browser, visit https://ondemand.pax.tufts.edu/
- Log in in with your Tufts credentials
- On the top grey menu bar, choose
Interactive Apps->IGV.
- Choose the following compute resource parameters:
- Number of house = 1
- Number of cores = 1
- Amount of Memory = 8GB
- Reservation = Bioinformatics Workshop
- Directory (leave blank)

-
Click the blue button
Launch IGVwhen it appears -
After this the IGV window will appear as a small window on a grey background.
-
Click the square icon in the top right corner to maximize the window.
Load reference genome
Choose reference genome by clicking the Genomes menu and selecting Load Genome from File...

- Navigate from your home directory to
hpcDay2/data/and select the fasta fileGCF_009858895.2_ASM985889v3_genomic.fna
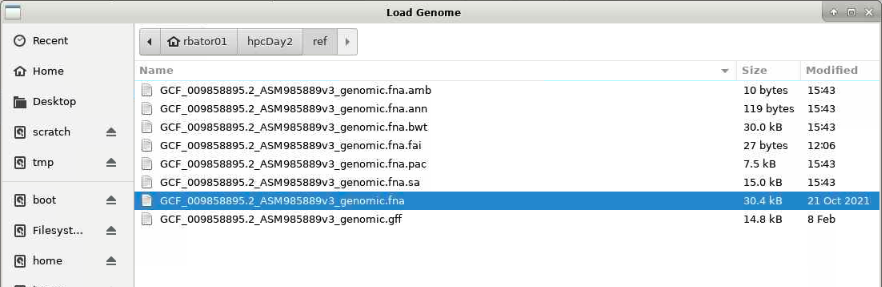
- Click
Open
You will see the name of the file and name of the sequence populate.
Load the GFF file
- Choose the GFF file by clicking on
Filemenu and selectingLoad from File...
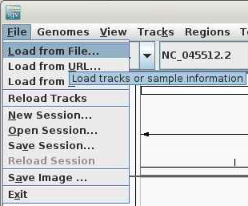
- As before, navigate to
tutln01/hpcDay2/data/and select the GFF fileGCF_009858895.2_ASM985889v3_genomic.gffand clickOpen
Load the BAM file
-
Choose the BAM file by clicking on
Filemenu and selectingLoad from File... -
Navigate to
tutln01/hpcDay2/results/and select the sorted BAM filesarscov2.srt.bam -
Click
Open -
It will take a minute or so to load all the reads. You can view progress in the lower right hand corner.
-
When it’s done, you will have the following view. Each row of data is called a track. There are four tracks visible: the top track shows the reference genome coordinates, followed by two tracks of our alignment (coverage and reads) followed by the GFF track showing the gene locations on our reference genome.
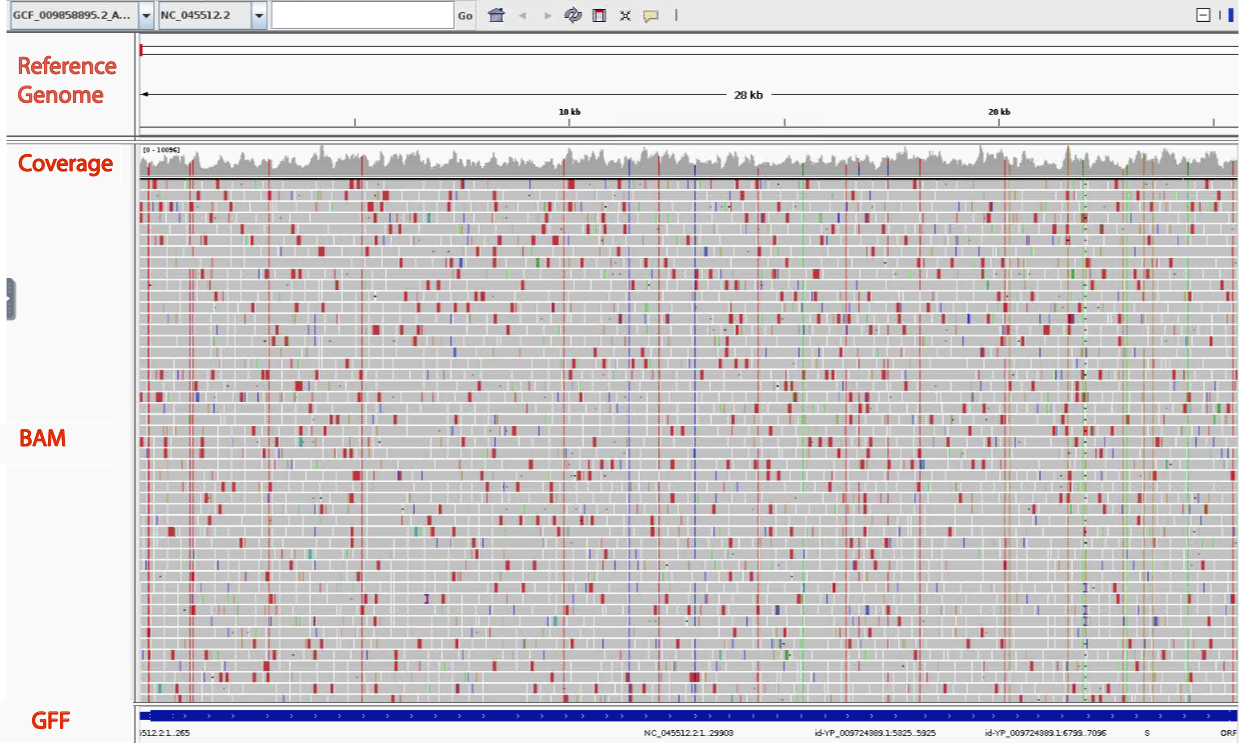
Examining a Gene
- Zoom in on the Gene that encodes the Spike protein by hovering with your mouse on the genome track and clicking and dragging over the portion that is directly above the “S” gene in the GFF track.

- We should see the coordinate box show roughly basepairs 22,000-25,000
NC_045512.2:22,000-25,000.

- Looking at the Coverage track we see there are five positions that are colored, which indicates a basepair mismatch with respect to the references sequence in over 20% of the reads a that position. we will examine one more closely.

Examining a Variant
-
This region contains one of the 4 mutations that differentiate the delta variant from the originally characterized sequence.
-
Select region around the variant at 22,995, or type
NC_045512.2:22,995into the search box. -
The result shows many short reads that are aligned to the reference but disagree with the reference nucleotide
Cinstead having anA. This corresponds to an amino acid change is a T>K change at protein position 478. We also see one read with a missing base at this position.

- We can get more information about the coverage and composition of that position by clicking ont the variant in the coverage track. We confirm that there are no reads that match the reference sequence, one
Gand one deletion.

Examining a Read
-
Similar to a variant, you can click on a read to get individual read information.
-
Reads can be colored in various ways. The default is to color by
Insert size and pair orientation. The insert size is the length in basepairs between sequencing adapters (which may be greater than the length of reads R1 + R2) and the order of mapping of R1 and R2. You can change this by right clicking on the alignment and choosingColor alignments by.
Note on IGV on demand for July 2022: I’ve observed behavior where the interface become no longer responsive to user actions. This likely means that more memory is needed. Sometimes it can be overcome by closing the browser tab and rejoining the session by clicking Launch IGV. The tech team is looking into this.