Approximate time: 20 minutes
Goals
- Connect to the HPC cluster via On Demand Interface
- Download data
Log into the HPC cluster’s On Demand interface
- Open a Chrome browser and enter the URL https://ondemand.cluster.tufts.edu
- Log in with your Tufts Credentials
- On the top menu bar choose
Clusters->Tufts HPC Shell Access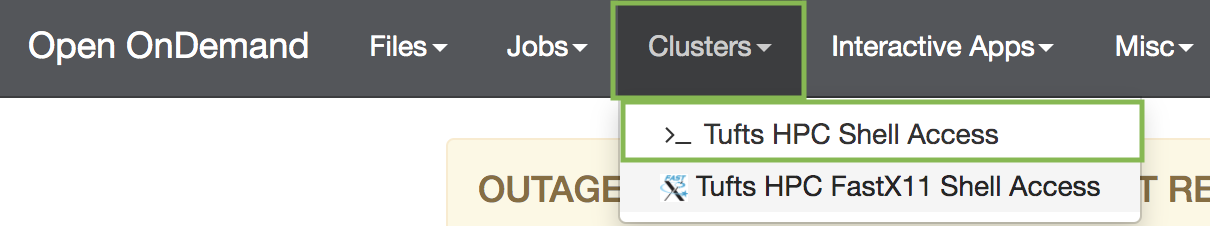
- Type your password at the prompt (the password will be hidden for security purposes):
tutln01@login.cluster.tufts.edu's password: - You’ll see a welcome message and a bash prompt, for example for user
tutln01:
[tutln01@login001 ~]$
This indicates you are logged in to the login node of the cluster.
- Type
clearto clear the screen
Set up for the analysis
Find 500M storage space
- Check how much available storage you have in your home directory by typing
showquota.
Result:
Home Directory Quota
Disk quotas for user tutln01 (uid 31394):
Filesystem blocks quota limit grace files quota limit grace
hpcstore03:/hpc_home/home
1222M 5120M 5120M 2161 4295m 4295m
Listing quotas for all groups you are a member of
Group: facstaff Usage: 16819478240KB Quota: 214748364800KB Percent Used: 7.00%
Under blocks you will see the amount of storage you are using, and under quota you see your quota.
Here, the user has used 1222M of the available 5120M and has enough space for our analysis.
- If you do not have 500M available, you may have space in a project directory for your lab.
These are located in
/cluster/tuftswith names like/cluster/tufts/labname/username/. If you don’t know whether you have project space, please email tts-research@tufts.edu.
Download the data
- Get an interaction session on a compute node (3 hours, 16 Gb memory, 4 cpu on 1 node) on the default partition (
batch) by typing:
srun --pty -t 3:00:00 --mem 16G -N 1 --cpus 4 bash
Notes:
If wait times are very long, you can try a different partitions by adding, e.g. -p preempt or -p interactive before bash.
If you go through this workshop in multiple steps, you will have to rerun this step each time you log in.
- Change to your home directory
cd
Or, if you are using a project directory:
cd /cluster/tufts/labname/username/
- Copy the course directory and all files in the directory (-R is for recursive):
cp -R /cluster/tufts/bio/tools/training/intro-to-ngs/ .
(Also available via: git clone https://gitlab.tufts.edu/rbator01/intro-to-ngs.git)
- Take a look at the contents using the
treecommand:
tree intro-to-ngs
You’ll see a list of all files
intro-to-ngs
├── all_commands.sh <-- Bash script with all commands
├── raw_data <-- Folder with paired end fastq files
│ ├── na12878_1.fq
│ └── na12878_2.fq
├── README.md <-- Contents description
└── ref_data <-- Folder with reference sequence
└── chr10.fa
2 directories, 5 files
Data for the class
Genome In a Bottle (GIAB) was initiated in 2011 by the National Institute of Standards and Technology “to develop the technical infrastructure (reference standards, reference methods, and reference data) to enable translation of whole human genome sequencing to clinical practice” (Zook et al 2012). We’ll be using a DNA Whole Exome Sequencing (WES) dataset released by GIAB for the purposes of benchmarking bioinformatics tools.

The source DNA, known as NA12878, was taken from a single person: the daughter in a father-mother-child ‘trio’. She is also mother to 11 children of her own, for whom sequence data is also available. (HBC Training). Father-mother-child ‘trios’ are often sequenced to study genetic links between family members.
As mentioned in the introduction, WES is a method to concentrate the sequenced DNA fragments in coding regions (exons) of the genome.
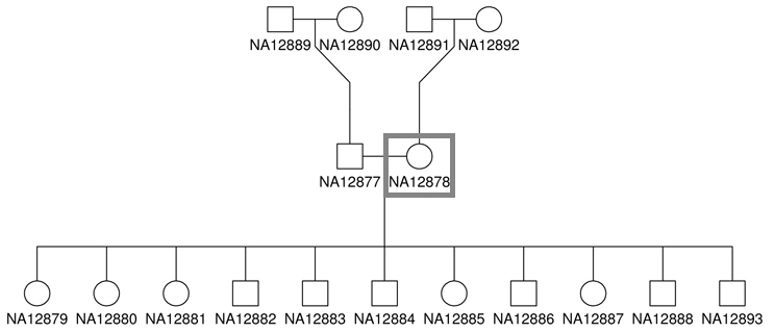
For this class, we’ve created a small dataset of reads that align to a single gene that will allow our commands to finish quickly.
Sample: NA12878
Gene: Cyp2c19 on chromosome 10
Sequencing: Illumina, Paired End, Exome