Obtaining the Reference data and NGS Sequencing data from public repositories
The US National Center for Biotechnology Information hosts repositories for many types of biomedical and genomics data. Today we’ll retrieve reference data from the Genomes Database FTP server as well as the Sequence Read Archive
Step 1: Galaxy Setup
Create a new history
- Click the + at the top of the history panel, on the right hand side of the screen
- To rename the history, click on the box Unnamed History, type ngs data workshop, and press enter
Step 2: Obtaining our Data
Our dataset is a SARS-CoV-2 Next Generation Sequencing sample. In this section we’ll obtain our reference data and our NGS reads in preparation for alignment.
Import the SARS-CoV2 genome and gene annotation from NCBI
- On the left hand side tool panel, click the Upload icon
- Click Paste/Fetch data
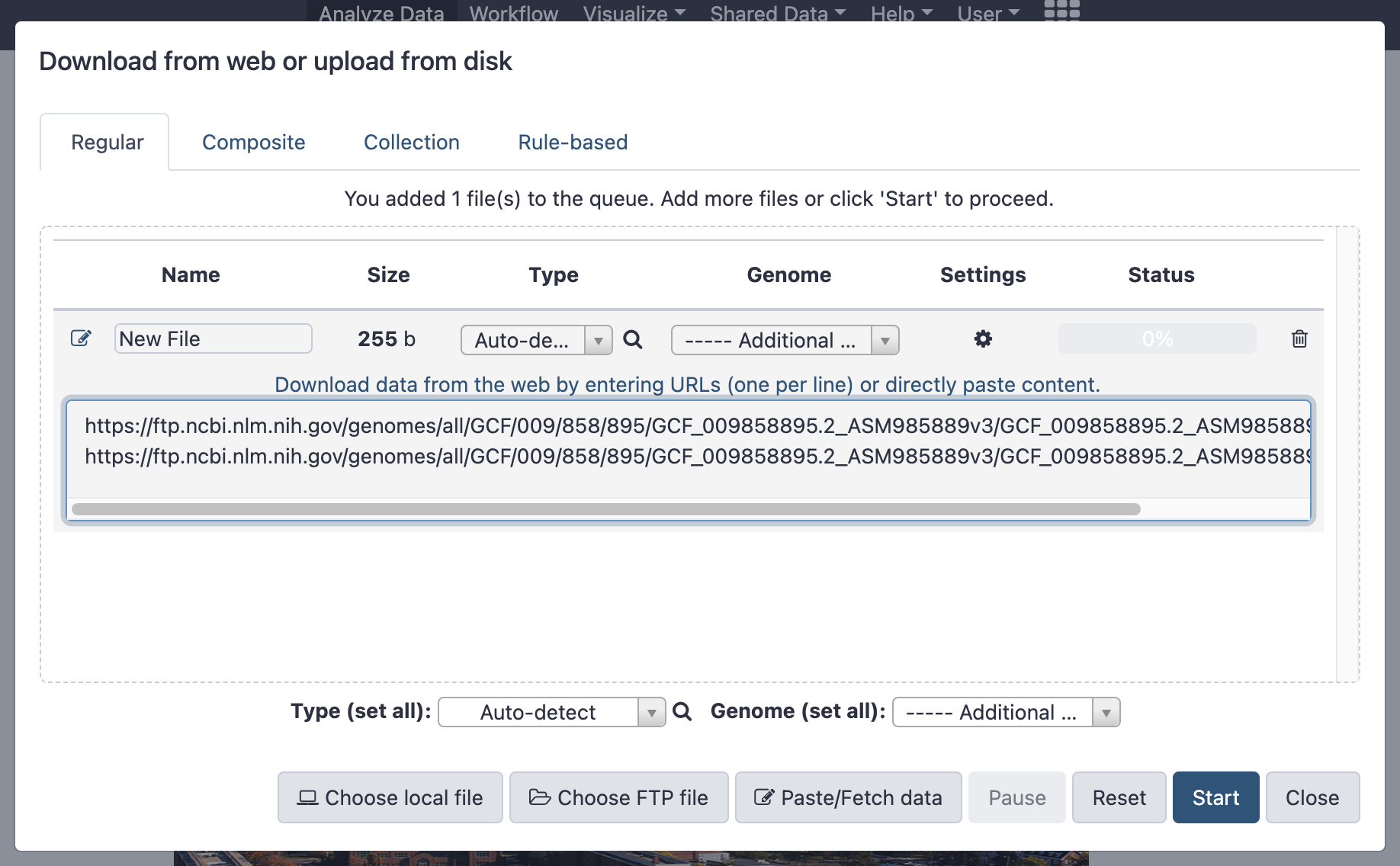
- Copy the following URLs and paste in the grey box:
https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/009/858/895/GCF_009858895.2_ASM985889v3/GCF_009858895.2_ASM985889v3_genomic.fna.gz
https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/009/858/895/GCF_009858895.2_ASM985889v3/GCF_009858895.2_ASM985889v3_genomic.gff.gz
- This is what you should see:
-
Click Start followed by Close
-
Two jobs will appear in the History, Grey (pending) -> Orange (running) -> Green (complete).
- Rename the genome file
- On the green box for the file ending in fna.gz, click on the

- Under Name, replace the URL in the name with “genome”
- Click Save
- On the green box for the file ending in fna.gz, click on the
- Rename the gene annotation file and decompress
- On the green box for the file ending in gff.gz, click on the
- Under Name, replace the URL in the name with “genes”
- Click Save
- Click on the Convert tab, and under Name select Convert compressed file to uncompressed
- Click Convert Datatype to generate a new dataset with the uncompressed file
- On the green box for the file ending in gff.gz, click on the
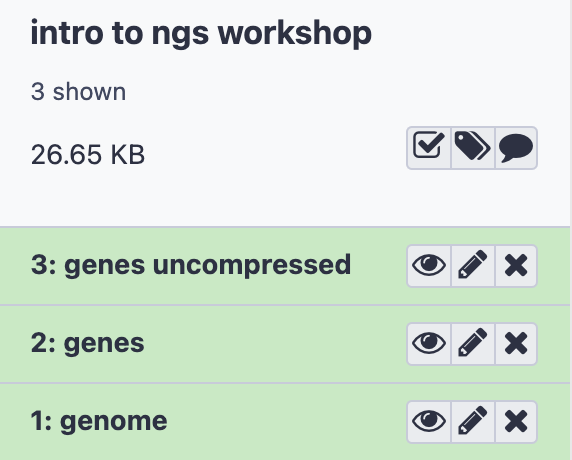
- Here is what you should see in your History
Fasta Format
The virus genome is in fasta format. Fasta format has two parts, a sequence identifier preceeded by a “>” symbol, followed by the sequence on subsequent lines. You can see a preview of it by clicking on the genome dataset in the History panel.

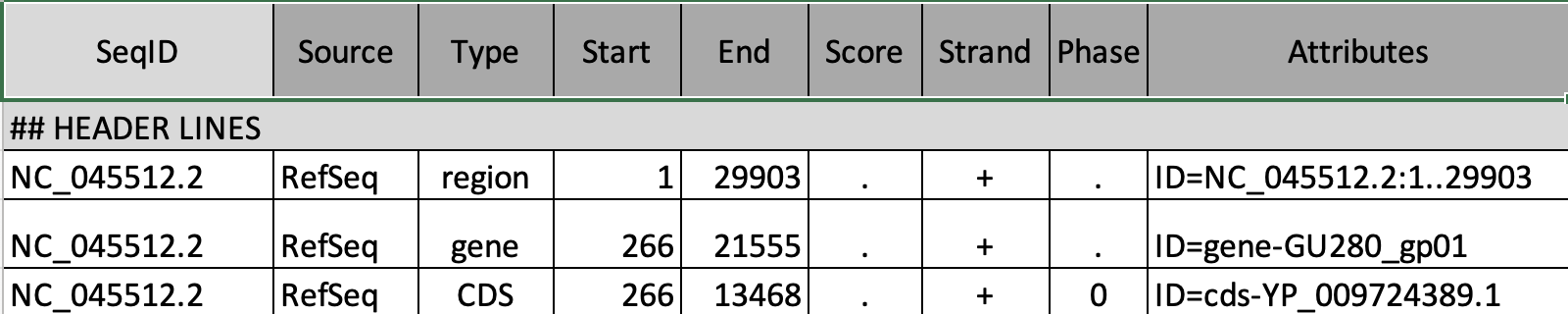
GFF Format
The gene annotation file is in Generic Feature Format (GFF). This formet tells us where genes are located in the reference genome.
To preview the GFF file, click on the ![]() on the genes uncompressed dataset.
Note that we must always be sure that our gene information and genome come from the same source.
on the genes uncompressed dataset.
Note that we must always be sure that our gene information and genome come from the same source.
Step 3: Import NGS sequencing data from Sequence Read Archive
We are interested in obtaining reads from the sample Viral genomic RNA sequencing of a B.1.617.2/Delta isolate; Severe acute respiratory syndrome coronavirus 2; RNA-Seq

Download Reads
We’ll download the data from Sequence Read Archive using a Galaxy tool called SRA Toolkit.
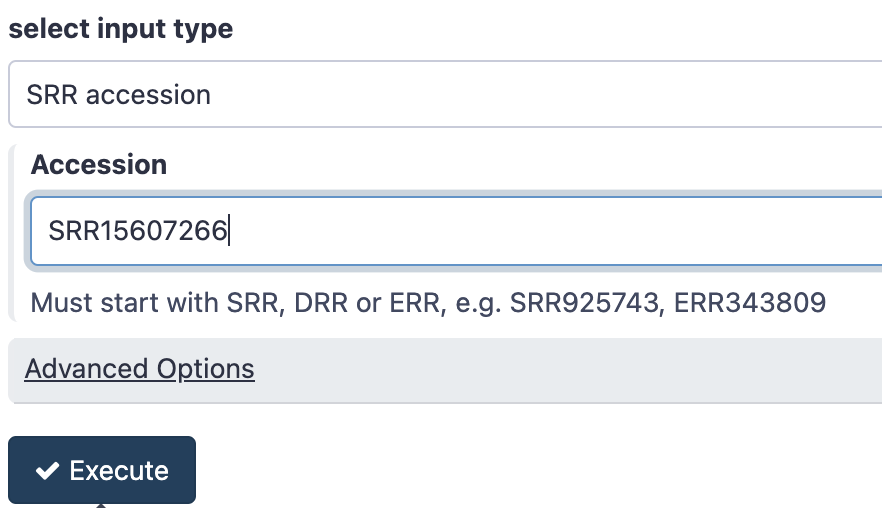
- In the Tool panel search box, search for a tool called “fasterq” and click on the tool under SRA toolkit called Faster Download and Extract Reads in FASTQ
- Under Accession paste the accession number
SRR15607266 - Click Execute
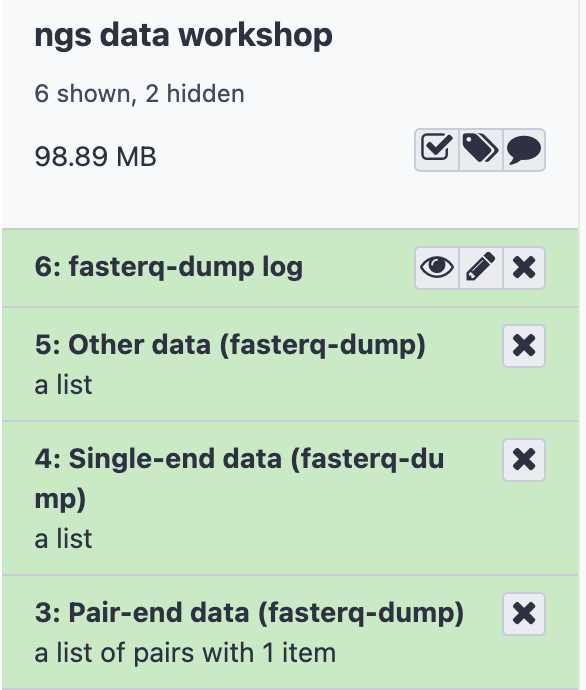
- The result will be four items in History
- Log: information about the total reads downloaded
- Other data: Empty, but can contain experiment metadata
- Single-end data: Empty, since this experiment has no single-end files
- Paired-end data: Two files, containing the forward and reverse reads for this sample
Fastq format
Fastq format is a way to store both sequence data and information about the quality of each sequenced position.
Each block of 4 lines contains one sequencing reads, for example:
@SRR15607266.1 1 length=76
NTTATCTACTTTTATTTCAGCAGCTCGGCAAGGGTTTGTTGATTCAGATGTAGAAACTAAAGATGTTGTTGAATGT
+SRR15607266.1 1 length=76
#8ACCGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG
- Sequence identifier
- Sequence
- + (optionally lists the sequence identifier again)
- Quality string
Paired end sequencing data will typically be stored as two fastq files, one for the forward and one for the reverse. Each file should contain the same number of reads, with the same labels, in the same order. If this convention is not followed, it could cause errors with downstream tools. Fortunately there are tools such as BBTools Repair that can help restore pairing information.
Base Quality Scores
The symbols we see in the read quality string are an encoding of the quality score:

A quality score is a prediction of the probability of an error in base calling:

Going back to our read, we can see that for most of our read the quality score is “G” –> “Q” = 38 -> Probability < 1/1000 of an error.
Preview Fastq data
- Click on the list Pair-end data (fasterq-dump) and the sublist SRR15607266 to expand the sample, you’ll see 2 sequencing files forward and reverse
- Click on the
 on the first sequence file forward and look at the fastq reads
on the first sequence file forward and look at the fastq reads