Approximate time: 20 minutes
Learning Objectives
- Sort and Index SAM/BAM files
- Mark duplicate reads in BAM file
Sort SAM file
Downstream applications require that reads in SAM files be sorted by reference genome coordinates (fields 3 and 4 in each line of our SAM file). This will assist in fast search, display and other functions.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|
| SRR098401.109756285 | 83 | chr10 | 94760653 | 60 | 76M | = | 94760647 | -82 | CTAA… | D?@A… |
We’ll use the Picard toolkit for this and othter SAM file manipulations.
Open another script in our course directory called picard.sh
cd ..
nano picard.sh
Enter the following text:
module load picard/2.8.0
picard SortSam \
INPUT=results/na12878.sam \
OUTPUT=results/na12878.srt.bam \
SORT_ORDER=coordinate
We have input our SAM file and we will output a Binary Alignment Map (BAM) file, which is a compressed version of SAM format.
Exit nano by typing ^X and follow prompts to save the file picard.sh.
To run the script:
sh picard.sh
Result:
[Fri May 08 15:38:55 EDT 2020] picard.sam.SortSam INPUT=results/na12878.sam OUTPUT=results/na12878.srt.bam SORT_ORDER=coordinate VERBOSITY=INFO QUIET=false VALIDATION_STRINGENCY=STRICT COMPRESSION_LEVEL=5 MAX_RECORDS_IN_RAM=500000 CREATE_INDEX=false CREATE_MD5_FILE=false GA4GH_CLIENT_SECRETS=client_secrets.json
[Fri May 08 15:38:55 EDT 2020] Executing as rbator01@pcomp31 on Linux 2.6.32-696.1.1.el6.x86_64 amd64; Java HotSpot(TM) 64-Bit Server VM 1.8.0_60-b27; Picard version: 2.8.0-SNAPSHOT
INFO 2020-05-08 15:38:56 SortSam Finished reading inputs, merging and writing to output now.
[Fri May 08 15:38:57 EDT 2020] picard.sam.SortSam done. Elapsed time: 0.02 minutes.
Runtime.totalMemory()=2058354688
Take a look at the results directory:
ls results
The result shows that the sorted BAM file has been created:
na12878.sam na12878.srt.bam
Mark Duplicates in BAM file
Many copies are made of a single DNA fragment during the sequencing process. The amount of duplication may not be the same for all sequences and this can cause biases in variant calling. Therefore, we mark the duplicates so the variant caller can focus on the unique reads.
Duplicate reads are identified based on their alignment coordinates and CIGAR string. For example, the below alignment appears to have a G to A mutation in the majority of reads:
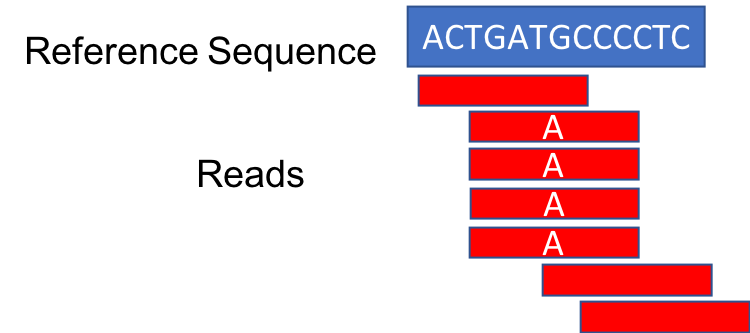
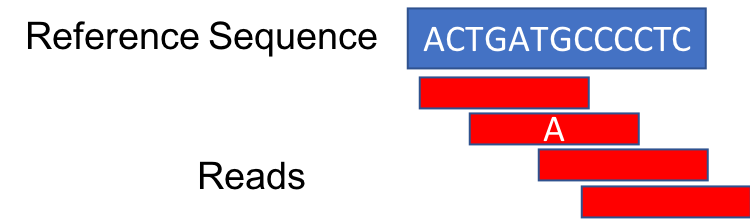
However, when the duplicates are removed, the number of reads supporting the mutation drops to one.
Let’s add this step to our picard.sh script in order to illustrate how to include multiple steps in a single script.
Note that when we run it, we’ll rerun our previous steps as well.
nano picard.sh
Add the following lines to the end of our script:
printf "..... Starting Mark Duplicates ....\n\n"
picard MarkDuplicates \
INPUT=results/na12878.srt.bam \
OUTPUT=results/na12878.srt.markdup.bam \
READ_NAME_REGEX=null \
METRICS_FILE=results/na12878.markdup.txt
The first line is a formatted print (printf) statement that will display useful log lines when our script is running.
The option READ_NAME_REGEX=null is added because our read names, downloaded from GIAB do not contain information about the position on the flowcell.
When present this information can help with estimating optical duplicated.
Typically, datasets do contain this information and it is best to omit this line when processing your data.
To run our script (Note this will rerun the first step as well. This is only for demonstration purposes. If you were developing this for your own use, you would instead write all commands and run the script once):
sh picard.sh
In addition to our previous log, we’ll see our log line, followed by the output from Mark Duplicates:
…
.... Starting Mark Duplicates ....
[Fri May 08 16:03:52 EDT 2020] picard.sam.markduplicates.MarkDuplicates INPUT=[results/na12878.srt.sam] OUTPUT=results/na12878.srt.markdup.sam METRICS_FILE=results/na12878.markdup.txt READ_NAME_REGEX=null MAX_SEQUENCES_FOR_DISK_READ_ENDS_MAP=50000 MAX_FILE_HANDLES_FOR_READ_ENDS_MAP=8000 SORTING_COLLECTION_SIZE_RATIO=0.25 REMOVE_SEQUENCING_DUPLICATES=false TAGGING_POLICY=DontTag REMOVE_DUPLICATES=false ASSUME_SORTED=false DUPLICATE_SCORING_STRATEGY=SUM_OF_BASE_QUALITIES PROGRAM_RECORD_ID=MarkDuplicates PROGRAM_GROUP_NAME=MarkDuplicates OPTICAL_DUPLICATE_PIXEL_DISTANCE=100 VERBOSITY=INFO QUIET=false VALIDATION_STRINGENCY=STRICT COMPRESSION_LEVEL=5 MAX_RECORDS_IN_RAM=500000 CREATE_INDEX=false CREATE_MD5_FILE=false GA4GH_CLIENT_SECRETS=client_secrets.json
[Fri May 08 16:03:52 EDT 2020] Executing as rbator01@pcomp31 on Linux 2.6.32-696.1.1.el6.x86_64 amd64; Java HotSpot(TM) 64-Bit Server VM 1.8.0_60-b27; Picard version: 2.8.0-SNAPSHOT
INFO 2020-05-08 16:03:52 MarkDuplicates Start of doWork freeMemory: 2042614304; totalMemory: 2058354688; maxMemory: 28631367680
INFO 2020-05-08 16:03:52 MarkDuplicates Reading input file and constructing read end information.
INFO 2020-05-08 16:03:52 MarkDuplicates Will retain up to 110120644 data points before spilling to disk.
INFO 2020-05-08 16:04:03 MarkDuplicates Read 9300 records. 0 pairs never matched.
INFO 2020-05-08 16:04:09 MarkDuplicates After buildSortedReadEndLists freeMemory: 2020313280; totalMemory: 2915041280; maxMemory: 28631367680
INFO 2020-05-08 16:04:09 MarkDuplicates Will retain up to 894730240 duplicate indices before spilling to disk.
INFO 2020-05-08 16:04:11 MarkDuplicates Traversing read pair information and detecting duplicates.
INFO 2020-05-08 16:04:11 MarkDuplicates Traversing fragment information and detecting duplicates.
INFO 2020-05-08 16:04:11 MarkDuplicates Sorting list of duplicate records.
INFO 2020-05-08 16:04:14 MarkDuplicates After generateDuplicateIndexes freeMemory: 3340626880; totalMemory: 10530324480; maxMemory: 28631367680
INFO 2020-05-08 16:04:14 MarkDuplicates Marking 864 records as duplicates.
WARNING 2020-05-08 16:04:14 MarkDuplicates Skipped optical duplicate cluster discovery; library size estimation may be inaccurate!
INFO 2020-05-08 16:04:14 MarkDuplicates Reads are assumed to be ordered by: coordinate
INFO 2020-05-08 16:04:16 MarkDuplicates Before output close freeMemory: 10507885056; totalMemory: 10530324480; maxMemory: 28631367680
INFO 2020-05-08 16:04:16 MarkDuplicates After output close freeMemory: 10507907720; totalMemory: 10530324480; maxMemory: 28631367680
[Fri May 08 16:04:16 EDT 2020] pic
Mark Duplicates Metrics file
The following is the metrics file na12878.markdup.txt generated by Picard Mark Duplicates:
| LIBRARY | UNPAIRED_READS_EXAMINED | READ_PAIRS_EXAMINED | SECONDARY_OR_SUPPLEMENTARY_RDS | UNMAPPED_READS | UNPAIRED_READ_DUPLICATES | READ_PAIR_DUPLICATES | READ_PAIR_OPTICAL_DUPLICATES | PERCENT_DUPLICATION | ESTIMATED_LIBRARY_SIZE |
| Unknown | 29 | 4620 | 2 | 35 | 14 | 425 | 0 | 0.093214 | 23546 |
Normal % duplication for exome sequencing data is 10-30%.
By scrolling to the left in this table we see that our percent duplication is 0.093214%.
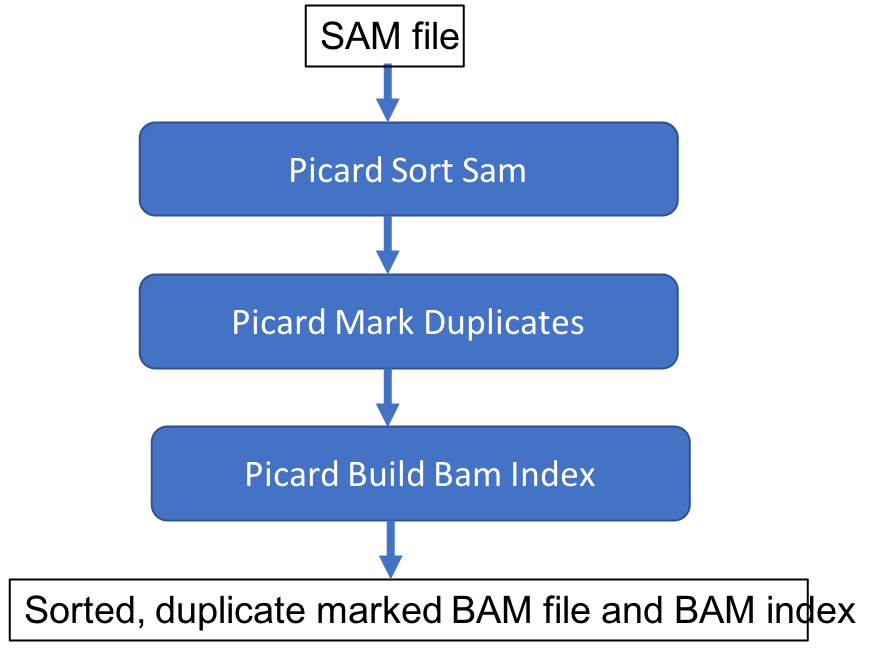
Index the BAM file
In order to view the alignment with the Integrated Genomics Viewer (IGV) we are required to create an index files for our BAM file. This facilitates fast lookup of genomics coordinates.
Let’s continue editing our script:
nano picard.sh
Add the following lines at the end of the script:
printf '.... Start BAM Indexing ....\n\n'
picard BuildBamIndex \
INPUT=results/na12878.srt.markdup.bam
Run our script:
sh picard.sh
Result, in addition to previous output:
.... Start BAM Indexing ....
[Fri May 08 16:24:17 EDT 2020] picard.sam.BuildBamIndex INPUT=results/na12878.srt.markdup.bam VERBOSITY=INFO QUIET=false VALIDATION_STRINGENCY=STRICT COMPRESSION_LEVEL=5 MAX_RECORDS_IN_RAM=500000 CREATE_INDEX=false CREATE_MD5_FILE=false GA4GH_CLIENT_SECRETS=client_secrets.json
[Fri May 08 16:24:17 EDT 2020] Executing as rbator01@pcomp31 on Linux 2.6.32-696.1.1.el6.x86_64 amd64; Java HotSpot(TM) 64-Bit Server VM 1.8.0_60-b27; Picard version: 2.8.0-SNAPSHOT
WARNING: BAM index file /cluster/home/rbator01/intro-to-ngs/results/na12878.srt.markdup.bai is older than BAM /cluster/home/rbator01/intro-to-ngs/results/na12878.srt.markdup.bam
INFO 2020-05-08 16:24:18 BuildBamIndex Successfully wrote bam index file /cluster/home/rbator01/intro-to-ngs/results/na12878.srt.markdup.bai
[Fri May 08 16:24:18 EDT 2020] picard.sam.BuildBamIndex done. Elapsed time: 0.01 minutes.
Runtime.totalMemory()=2058354688
We can see the files that were generated by typing ls results
na12878.sam
na12878.srt.bam
na12878.srt.markdup.bam
na12878.markdup.txt
na12878.srt.markdup.bai <--- Index file
BAM Visualization with IGV
- With a Chrome web browser, visit https://ondemand.cluster.tufts.edu
- Login with your Tufts credentials
-
Choose Interactive Apps->IGV. Set parameters, click “Launch”
- Choose
Interactive Apps->IGV. Set parameters below and , clickLaunch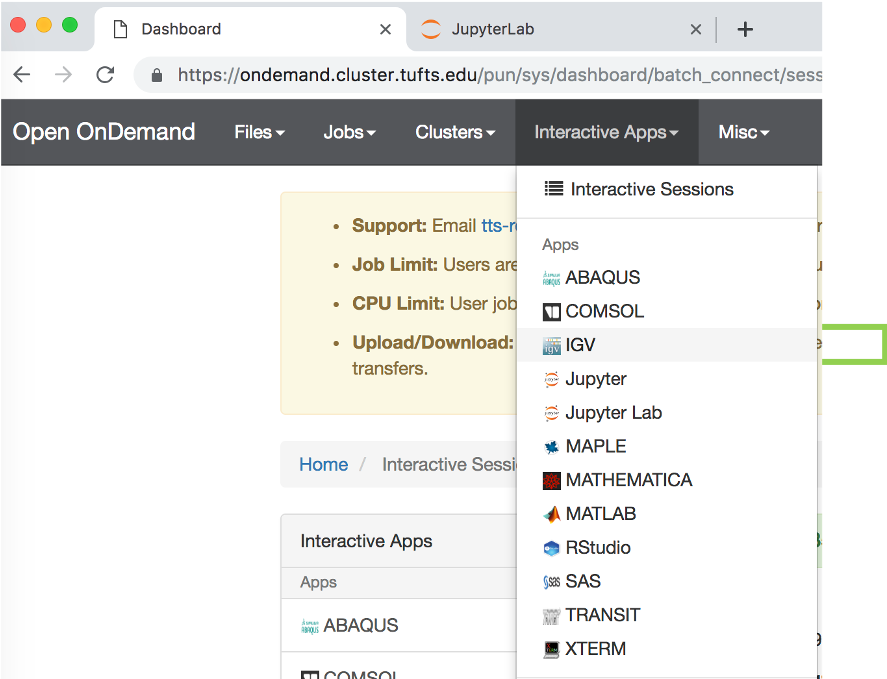
- Choose the following compute resource parameters: 1 hour, 2 cores, 4 GB memory, Default Batch Parition, Default Reservation
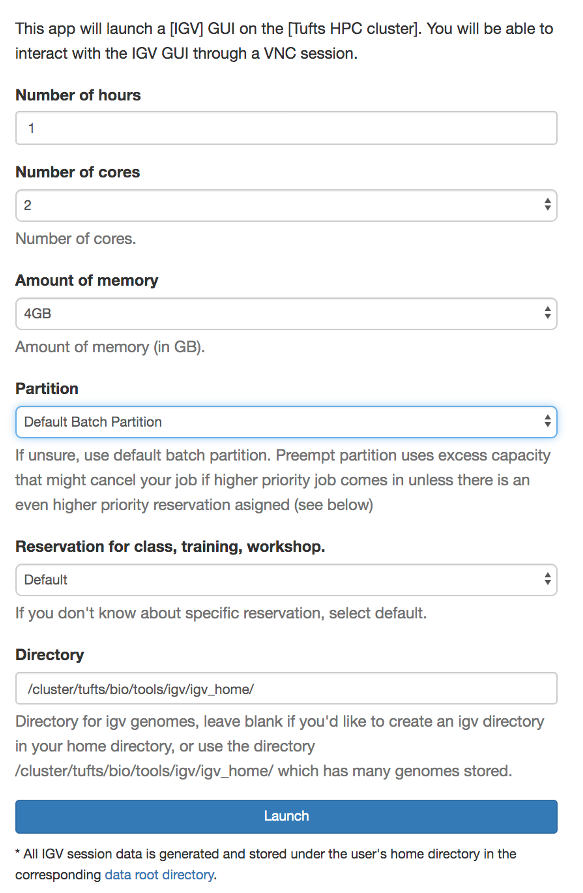
- Click the blue button
Launch NoVNC in New Tabwhen it appears
After this the IGV window will appear, probably as a small window on a grey background. Click the square icon in the top right corner to maximize the window.
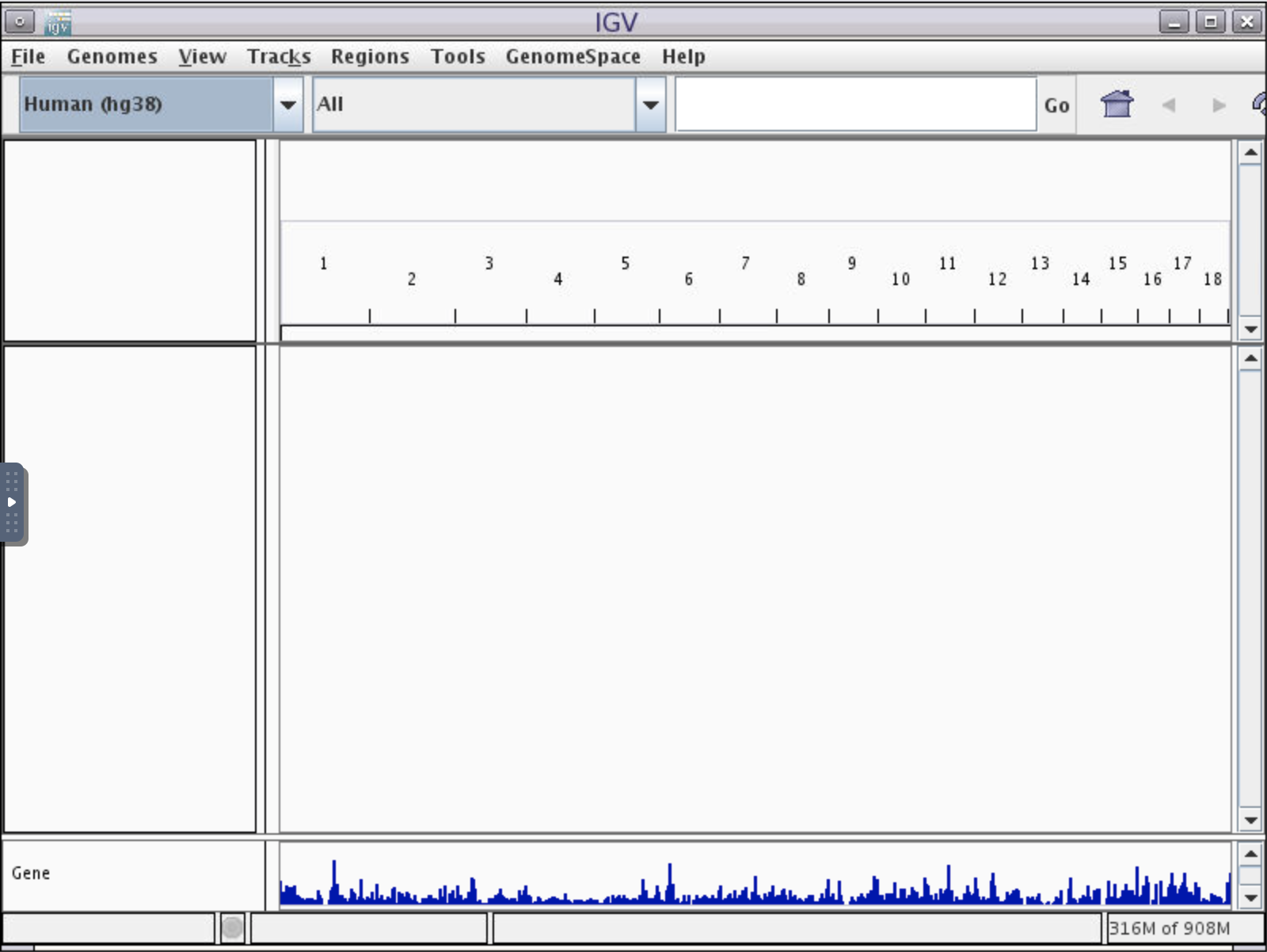
Load reference genome and BAM file
- Choose reference genome by clicking the
Genomesmenu and selectingLoad Genome from Server...
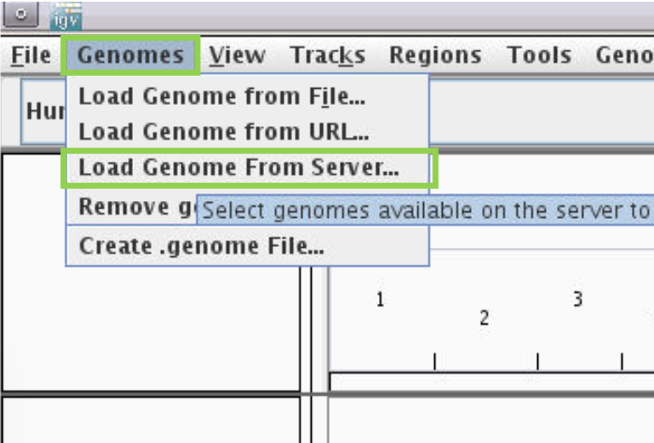
- Scroll down to
Human hg38
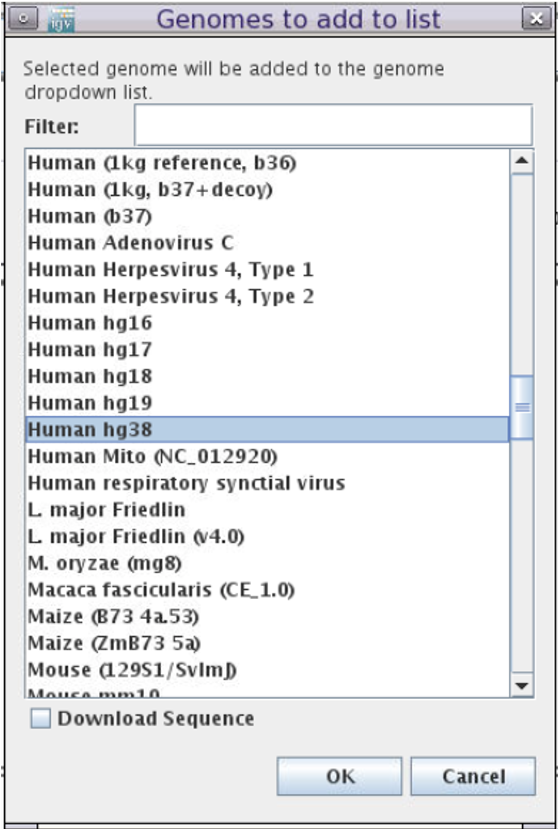
-
DO NOT check
Download Sequnence -
Click
OK -
Load the BAM file by clicking the
Filemenu and selectLoad from File...
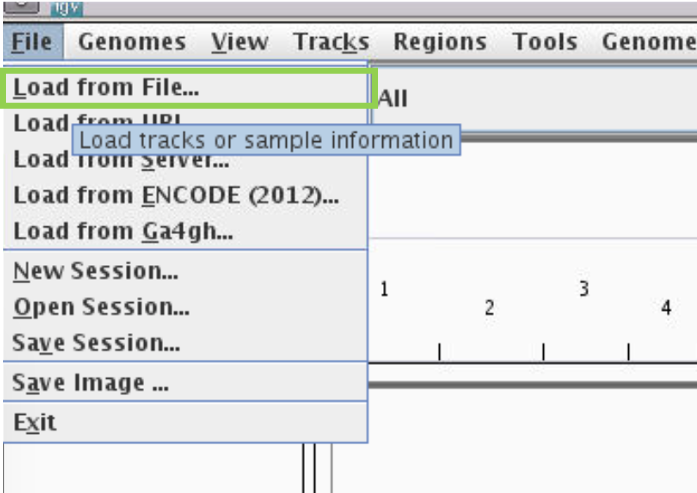
-
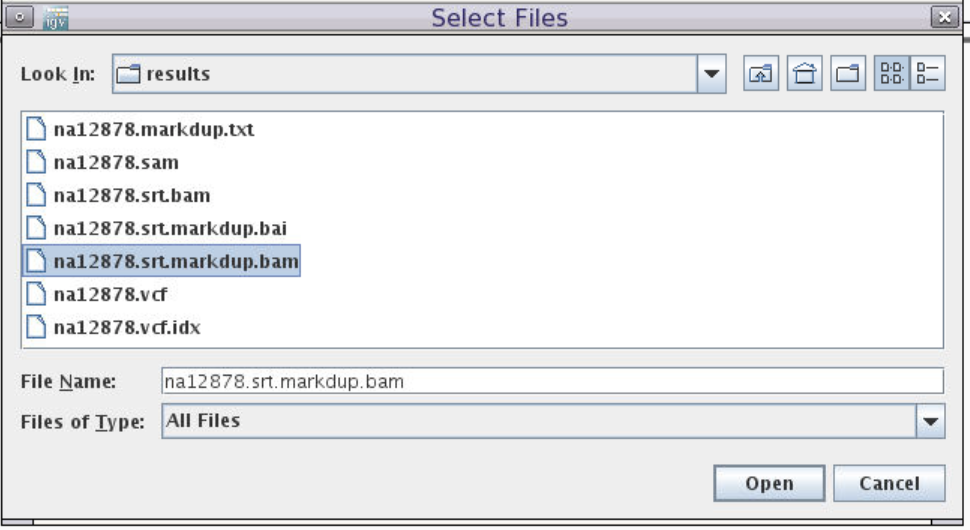
Navigate to the results folder in the course directory, e.g.
/cluster/home/your-user-name/intro-to-ngs/results. -
Select
na12878.srt.markdup.bam
You will have the following view:
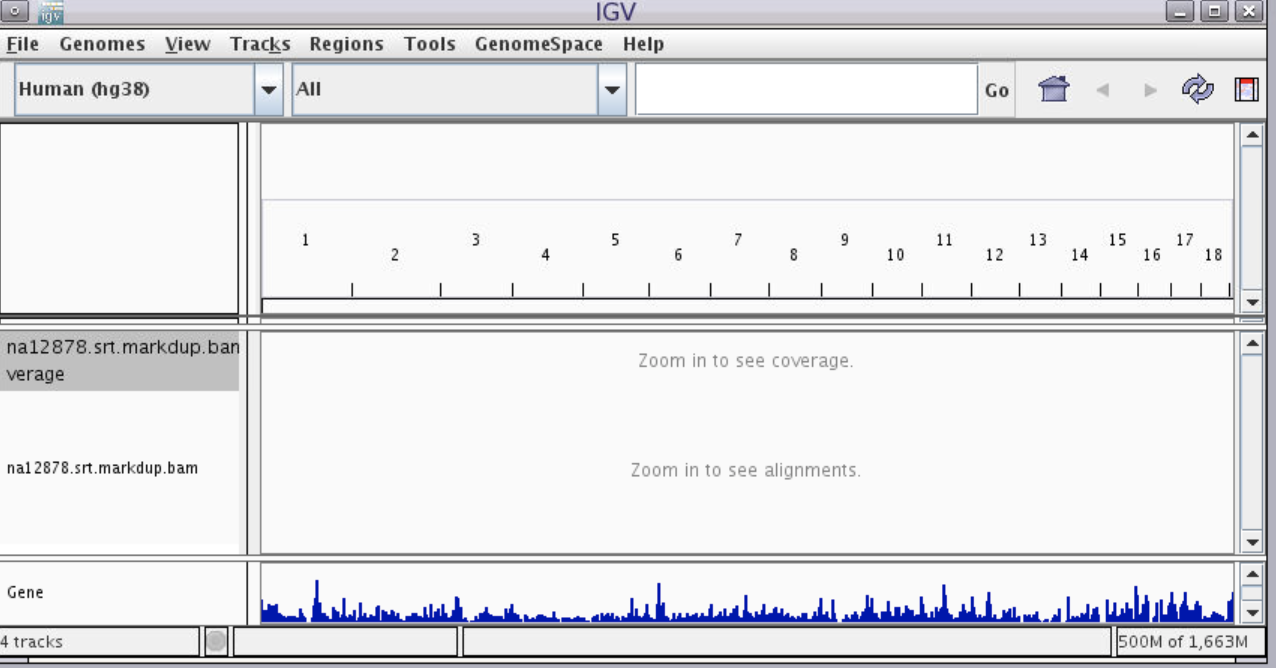
Each row of data is called a track. There are five tracks visible: the top track shows the pq bands of the entire chromosome, followed by the reference genome coordinate track, followed by two tracks of our alignment (coverage and reads, respectively) which don’t yet show data, followed by a reference genome annotation track called “Genes”.
Examining a gene
- In the box indicated in green below, type gene name “Cyp2c19” and hit enter. You will see the gene model display in the “Genes” track, showing vertical bars where exons are located
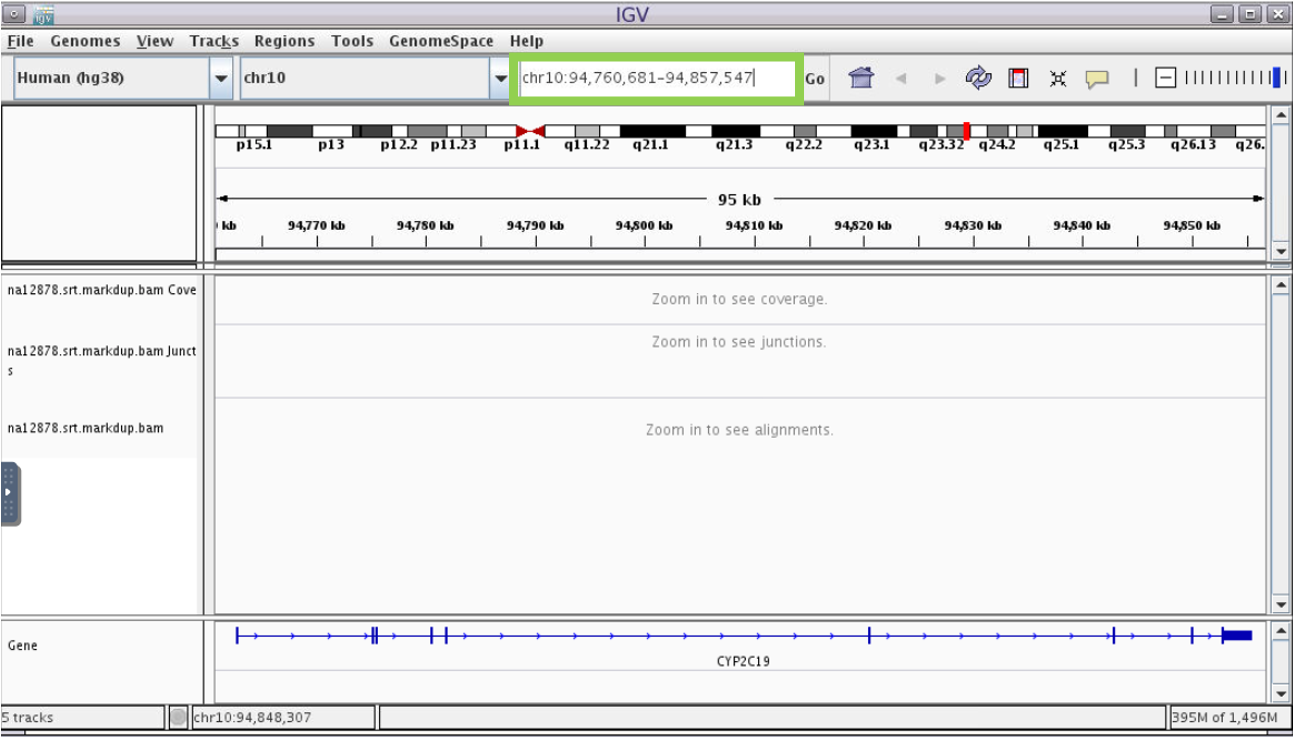
Troubleshooting tip: At times IGV on demand will stop allowing the user to type input.
If that happens, close the tab, go back to the on demand window, rejoin the session by clicking Launch NoVNC in New Tab.
- Let’s zoom in on exon 7. You can hover over exons in the
Genestrack to get information such as exon number. Click and drag over a region in the reference coordinate track to zoom in on exon 7 (highlighted in green below.)
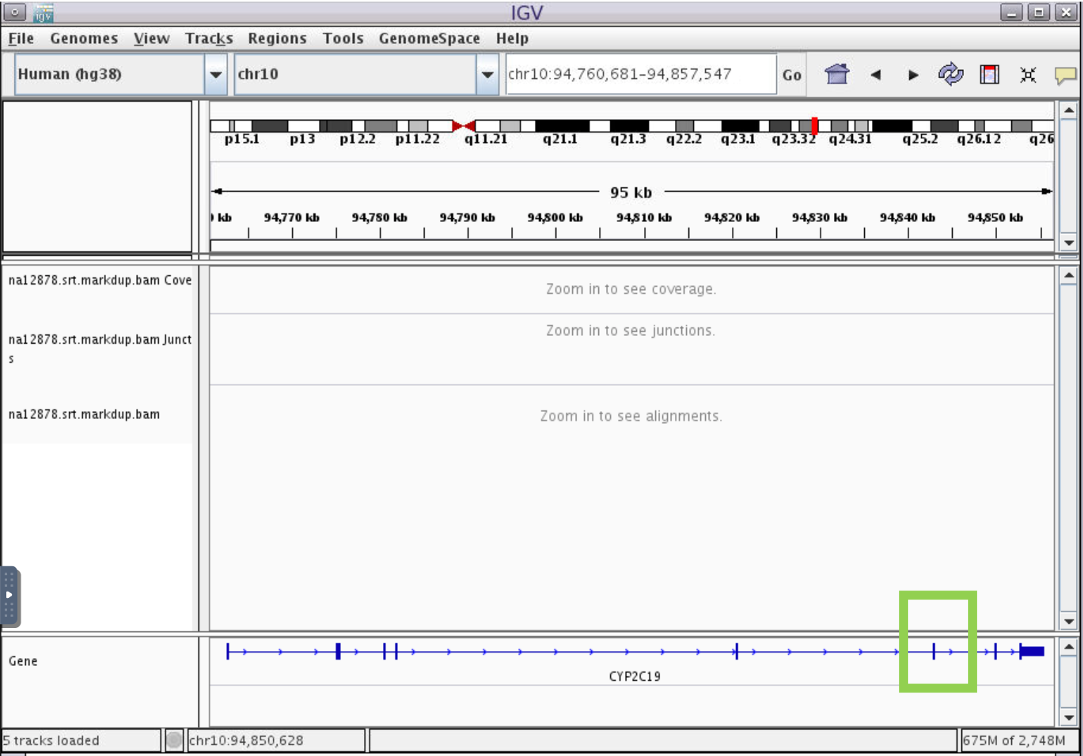
- We can see that there is a variant in this exon.
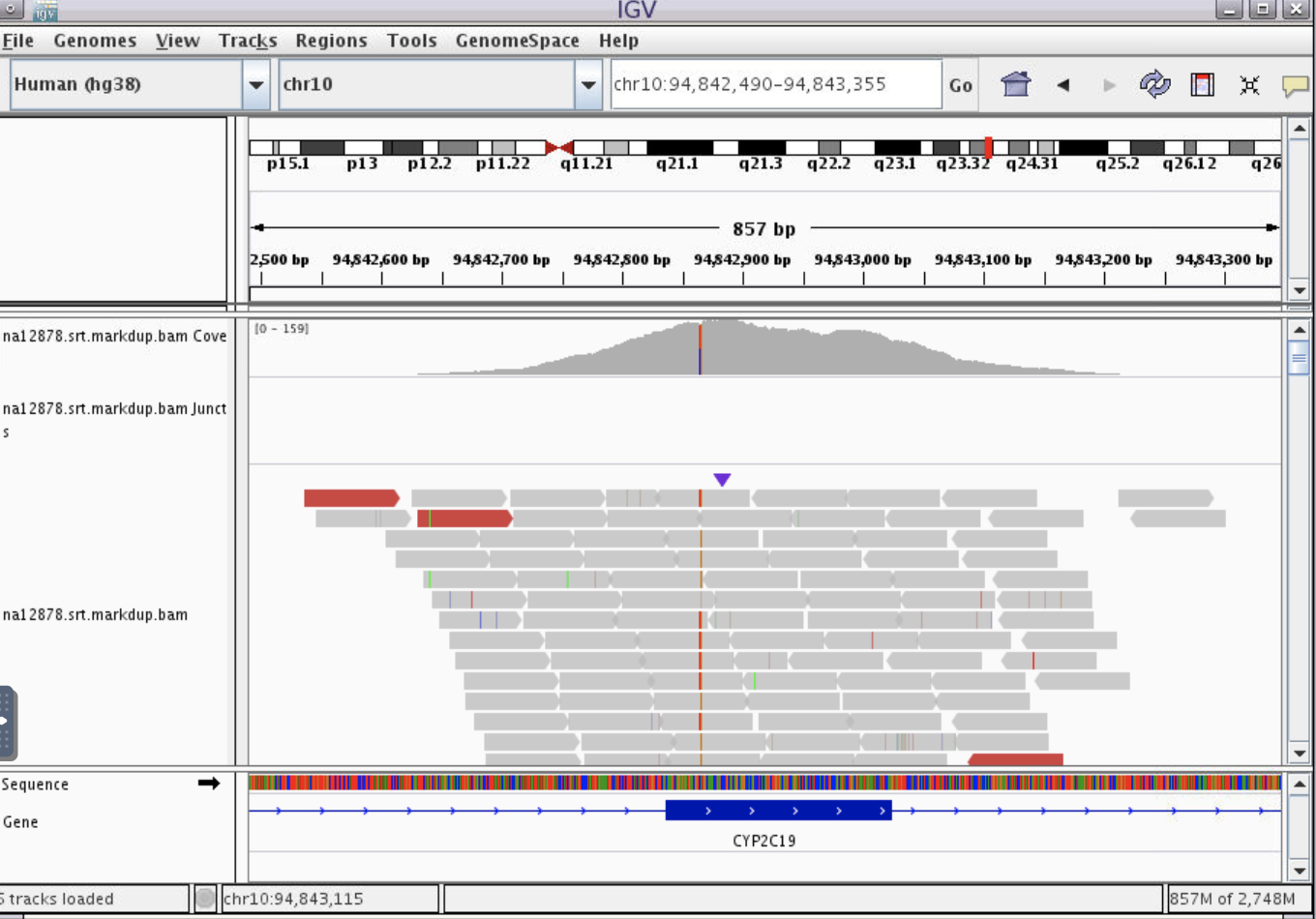
- Zoom in even further until the nucleotide letters are clear. Then, hover with your mouse over the coverage track to find out more information about this variant.
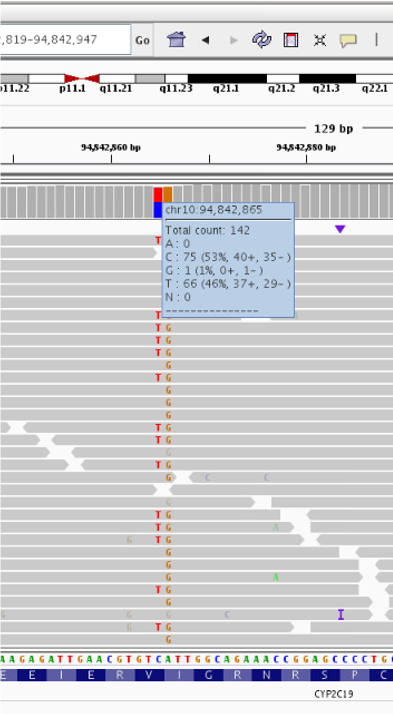
It appear there are two variants next to each other: heterozygousC>T at position chr10:94,842,865 and homozygous
A>G at position chr10:94,842,866. Next, we’ll explore the meaning of these variants.
This lesson adapted from HBC NGS Data Analysis